


Abstract
TENB2, also known as tomoregulin or transmembrane protein with epidermal growth factor–like and 2 follistatin-like domains, is a transmembrane proteoglycan overexpressed in human prostate tumors. This protein is a promising target for antimitotic monomethyl auristatin E (MMAE)–based antibody–drug conjugate (ADC) therapy. Nonlinear pharmacokinetics in normal mice suggested that antigen expression in normal tissues may contribute to targeted mediated disposition. We evaluated a predosing strategy with unconjugated antibody to block ADC uptake in target-expressing tissues in a mouse model while striving to preserve tumor uptake and efficacy. Methods: Unconjugated, unlabeled antibody was preadministered to mice bearing the TENB2-expressing human prostate explant model, LuCaP 77, followed by a single administration of 111In-labeled anti-TENB2-MMAE for biodistribution and SPECT/CT studies. A tumor-growth-inhibition study was conducted to determine the pharmacodynamic consequences of predosing. Results: Preadministration of anti-TENB2 at 1 mg/kg significantly increased blood exposure of the radiolabeled ADC and reduced intestinal, hepatic, and splenic uptake while not affecting tumor accretion. Similar tumor-to-heart ratios were measured by SPECT/CT at 24 h with and without the predose. Consistent with this, the preadministration of 0.75 mg/kg did not interfere with efficacy in a tumor-growth study dosed at 0.75 mg or 2.5 mg of ADC per kilogram. Conclusion: Overall, the potential to mask peripheral, nontumor antigen uptake while preserving tumor uptake and efficacy could ameliorate toxicity and may significantly affect future dosing strategies for ADCs.
Antibody–drug conjugates (ADCs) hold promise as enhanced antibody therapeutics by combining the antigen specificity of antibodies with the potency of cytotoxic drugs (1,2). The success of ADC therapeutics hinges on the balance between specific delivery of toxic chemotherapeutics to tumors and minimizing the risk of side effects to normal tissues (3). Toxicity in ADC therapies may result from premature systemic release of toxin or from antigen-mediated ADC uptake in normal tissues. The former may be addressed through optimization of linker stability, whereas the latter is often dictated by target selection. An ideal ADC target is exclusively expressed on tumor cells. However, many validated targets for solid tumors also exhibit low expression in normal tissues. Because of the potency of the drug component, ADCs with high uptake in nontumor tissues may have limited therapeutic windows (4).
A potential means of decreasing antibody uptake in nontumor tissues involves blockage of binding sites by predosing with an unconjugated antibody, which is typically less potent or inert (5). This strategy has been successfully applied in nuclear imaging and radioimmunotherapy (6–8). Predosing often improves biodistribution into the effect compartment, yielding improved tumor accumulation of radioimmunoconjugates in xenograft models and in the clinic (9–11). Herein, we pursue a similar predosing strategy to block normal murine tissue uptake of an ADC in a prostate cancer model without compromising tumor uptake.
TENB2, also known as tomoregulin or transmembrane protein with epidermal growth factor–like and 2 follistatin-like domains, is a proteoglycan overexpressed in human prostate tumors (12) and is being pursued as an ADC target (13). Although its normal biologic function remains elusive, elevated TENB2 expression has been associated with higher prostate cancer grade and hormone independence (14). Thio-anti-TENB2-MC-vc-PAB-MMAE (designated henceforth as anti-TENB2-MMAE) is a humanized anti-TENB2 ThioMab(15) conjugated to a potent antimitotic auristatin drug, monomethyl auristatin E (MMAE), through a maleimido-caproyl-valine-citrulline-para-amino-benzyloxy carbonyl (MC-vc-PAB) linker designed to be cleaved by lysosomal proteases (16). An ideal target, TENB2 exhibits limited expression in healthy human tissues, with positive immunostaining confined to the central nervous system tissues and normal prostate (13). Positive staining in murine brain, but not prostate, was also reported; however, the authors did not specify whether other mouse tissues were tested (13). A saturable antigen sink for anti-TENB2-MMAE causes nonlinear pharmacokinetics and pronounced target-mediated clearance in mice and rats (17), thus providing an ideal preclinical model for ADCs having undesirable peripheral target antigen expression. In a related but separate study (18), we carefully examined the prevalence and level of TENB2 expression in non–tumor-bearing mice by pharmacokinetics, competitive tissue uptake, and immunohistochemistry and confirmed expression and significant intestinal uptake of the anti-TENB2 ADC.
We hypothesized that preadministration of unconjugated anti-TENB2 antibody at a suitable dose level would saturate the low-to-moderate peripheral antigen expression while maintaining tumor uptake in a TENB2-overexpressing explant model. Blood pharmacokinetics, tissue distribution, and imaging data were generated and collectively used to delineate the relationships among predose level, nontumor tissue uptake, and tumor uptake. In addition, an in vivo tumor-growth-inhibition study was conducted to determine the pharmacodynamic consequences of predosing.
MATERIALS AND METHODS
Reagents
Cysteine residues were engineered at Ala114 positions of the heavy chains to produce the ThioMab variant of anti-TENB2 (15). An anti–6-transmembrane epithelial antigen of prostate 1 (STEAP1) ThioMab was also constructed as an IgG1 control antibody. Partial reduction and reoxidation yielded 2 free thiol groups per antibody, which were conjugated to MMAE via the protease-labile MC-vc-PAB linker. The drug-to-antibody ratio of ADCs was determined to be approximately 2 by liquid chromatography mass spectrometry analysis.
Radiochemistry
Anti-TENB2-MMAE was labeled with DOTA for 111In complexation by random modification of lysine residues as previously described (19). We observed 2.9 and 3.1 DOTA units, respectively, by reduced and intact liquid chromatography mass spectrometry. Radiometric measurement of the average number of DOTA chelates attached per antibody molecule gave a value of 3.7 for DOTA-anti-TENB2-MMAE. The DOTA conjugate showed acceptable (74%) retention of antigen binding, measured as percentage recovery in a TENB2-specific total antibody enzyme-linked immunosorbent assay that used TENB2 antigen as the capture reagent and antihuman Fc conjugated to horseradish peroxidase for detection. The assay has a lower limit of quantification of 8.2 ng/mL with a minimum dilution of 1:100. Incorporation of 111In into DOTA-anti-TENB2-MMAE was achieved with an 84% radiochemical yield, yielding a specific activity of 0.61 MBq/μg using reported procedures (19). Size-exclusion high-performance liquid chromatography demonstrated greater than 95% radiochemical purity for all radioimmunoconjugates.
LuCaP 77 Explant Mouse Model
The explant model of prostate cancer, LuCaP 77, was obtained from the University of Washington. The LuCaP 77 explant model was maintained by serial implantations in male nude mice for 2 passages, then in C.B-17 Fox Chase severe combined immune deficient (SCID) mice for 21 passages at the University of Washington and subsequently in male C.B-17 SCID beige mice (CB17.Cg-PrkdcscidLystbg/Crl) from Charles River Laboratories for continued passages at Genentech. When donor mice had tumors of between 800 and 1,000 mm3, tumor tissue was aseptically removed and dissected into small implantable-sized pieces (~20−30 mm3), which were subcutaneously implanted into the right flanks of male C.B-17 SCID beige mice, followed by skin closure using wound clips.
Flow Cytometry
Cross-reactivity of anti-TENB2 to human and mouse TENB2 was evaluated in human embryonic kidney cells (HEK293; American Type Culture Collection) that were stably transfected with human or mouse TENB2. Cells were selected and maintained using G418 antibiotic solution (Invitrogen) at 400 μg/mL, grown to 90% confluence, and removed from plates using cell dissociation buffer (Invitrogen). Cells were washed and resuspended in fluorescence-activated cell-sorting buffer (phosphate-buffered saline with 1% bovine serum albumin) and incubated for 45 min with humanized anti-TENB2 (0.6 μg/mL), followed by a 30-min incubation with antihuman secondary antibody conjugated to phycoerythrin. Analysis was performed with a FACS Caliber flow cytometer (BD Biosciences).
The binding of the anti-TENB2 antibody to its antigen was evaluated in a TENB2-transfected human prostate carcinoma cell line (PC3; American Type Culture Collection). Briefly, PC3 cells were transfected with TENB2, and PC3-TENB2 stable lines were generated, selected, and maintained with G418 at 100 μg/mL, with cell treatment and analysis by flow cytometry similar to that for HEK293 cells.
The expression of TENB2 in the LuCaP 77 model was also evaluated by flow cytometry. Briefly, LuCaP 77 tumors were harvested and treated with 1% bovine serum albumin in phosphate-buffered saline with a collagenase enzymatic mixture. Tumors were incubated for 15 min at 37°C, and cells were strained and washed in phosphate-buffered saline containing 1% bovine serum albumin. Cells were spun down and resuspended in the ammonium chloride–potassium lysis buffer to lyse the red blood cells at 4°C. Cells were treated in a manner similar to HEK293 and PC3 cells and analyzed by flow cytometry.
Dose-Escalation Study
All in vivo protocols, housing, and anesthesia were approved by the Institutional Animal Care and Use Committees of Genentech Laboratory Animal Resources, in compliance with the regulations of the Association for Assessment and Accreditation of Laboratory Animal Care. Male SCID beige mice ranging from 6 to 8 wk old and weighing 20–30 g received subcutaneously implanted LuCaP 77 prostate tumor explants, which reached 250–315 mm3 in 52 d.
The dose-escalation study was conducted with 111In-anti-TENB2-MMAE tracer at 0.83 MBq/μg, resulting in a 0.01-mg dose per kilogram at the tracer-only level after predosing at 0, 1, 3, or 10 mg of anti-TENB2 per kilogram. Under ketamine–xylazine anesthesia, whole blood was collected in lithium heparin tubes at 4 h and 1, 2, and 3 d. Tissues were harvested at 1 and 3 d after dosing (3 mice per group) for analysis as previously described (20).
SPECT/CT
SPECT/CT of mice with 111In-anti-TENB2-MMAE was performed as an adjunct to the biodistribution study. Radiolabeling involved incubation of 496.5 MBq of 111InCl3 and 462 μg of DOTA-ADC, and final specific activity of the tracer was 0.61 MBq/μg. Selected mice received pretreatment (10 mg/kg) of unlabeled, non–drug-conjugated anti-TENB2 ThioMab or IgG1 control (anti-STEAP1) ThioMab at 24 h before injection of the radiotracer. All mice received 45.9–49.2 MBq of 111In-anti-TENB2-MMAE (3.0–3.2 mg/kg at tracer only), and SPECT/CT was performed as previously reported (20). Tracer specific activities used in imaging and dose-escalation studies were similar (0.61 and 0.83 MBq/μg, respectively). Mice were subsequently euthanized under sedation and tissues collected for γ-counting in a manner identical to that used for the nonimaging arm of the study.
Efficacy Study
The LuCaP 77 explant model was used to evaluate the tumor-growth inhibition of anti-TENB2-MMAE, which was administered as a single agent and as a single dose with or without a predose of unconjugated anti-TENB2. When tumors reached a volume in the range of 111−252 mm3, animals were randomized into groups of 8 mice each and received an intravenous bolus injection of test materials. Mice received histidine buffer as a vehicle control, 0.75 or 2.5 mg of anti-TENB2-MMAE per kilogram only, or a predose of 0.75 mg of anti-TENB2 per kilogram 24 h before the dose of 0.75 or 2.5 mg/kg of anti-TENB2-MMAE. Mice were euthanized before tumors became ulcerated or reached the maximum allowable volume (3,000 mm3).
Pharmacokinetic Exposure in Efficacy Study
After a single intravenous dose, sparse pharmacokinetic samples of plasma were collected on days 2, 4, 8, and 15 and analyzed for the concentrations of conjugated antibody bearing at least one cytotoxic drug. This conjugated antibody enzyme-linked immunosorbent assay uses anti-MMAE antibody (Seattle Genetics Inc.) as a capture reagent and a biotinylated antigen, followed by streptavidin–horseradish peroxidase for detection. The assay has a lower limit of quantification of 1.3 ng/mL, with a minimum dilution of 1:100.
RESULTS
Tumor Model Characterization
The LuCaP 77 tumor–bearing mouse model was characterized for its utility in evaluating antigen-mediated anti-TENB2-MMAE uptake in both tumor and murine tissue. Although other tumor models have demonstrated expression of TENB2, we selected this model because of its high expression level and because it was derived from femur metastases of a hormone-refractory prostate cancer patient whose resistance to chemotherapy would suggest a need for targeted approaches such as ADCs (21,22). Anti-TENB2 showed similar binding to both human and murine TENB2 expressed in stably transfected HEK293 cells (Figs. 1A and 1B, respectively). The demonstration of species cross-reactivity was essential in validating our animal model, consisting of a TENB2-expressing human explant tumor growing in a mouse host whose normal tissues express the same antigen. To confirm the surface expression of TENB2 in the LuCaP 77 model, we established the binding of anti-TENB2 antibody to a TENB2-transfected PC3 cell line. Figure 1C shows that anti-TENB2 antibody bound to the surface of PC3 cells transfected with TENB2 with high affinity. Protein expression of both TENB2 and STEAP1 in the LuCaP 77 model was subsequently demonstrated through flow cytometry on cells obtained from subcutaneous LuCaP 77 xenografts (Fig. 1D).

Flow cytometry analysis demonstrates cross-reactive binding of anti-TENB2 to human and murine TENB2 and TENB2 expression in LuCaP 77 explant model. Anti-TENB2 antibody showed similar binding to HEK293 cell line that was stably transfected with both human (A) and murine (B) TENB2. Both anti-TENB2 and anti-STEAP1 antibodies, but not antiphycoerythrin control, show cell surface protein expression in cells from PC3 cells stably transfected with TENB2 (C) and in cells derived from LuCaP 77 explant model (D). FL2 = fluorescence channel 2; Max = maximum; PE = phycoerythrin.
Dose-Escalation Study
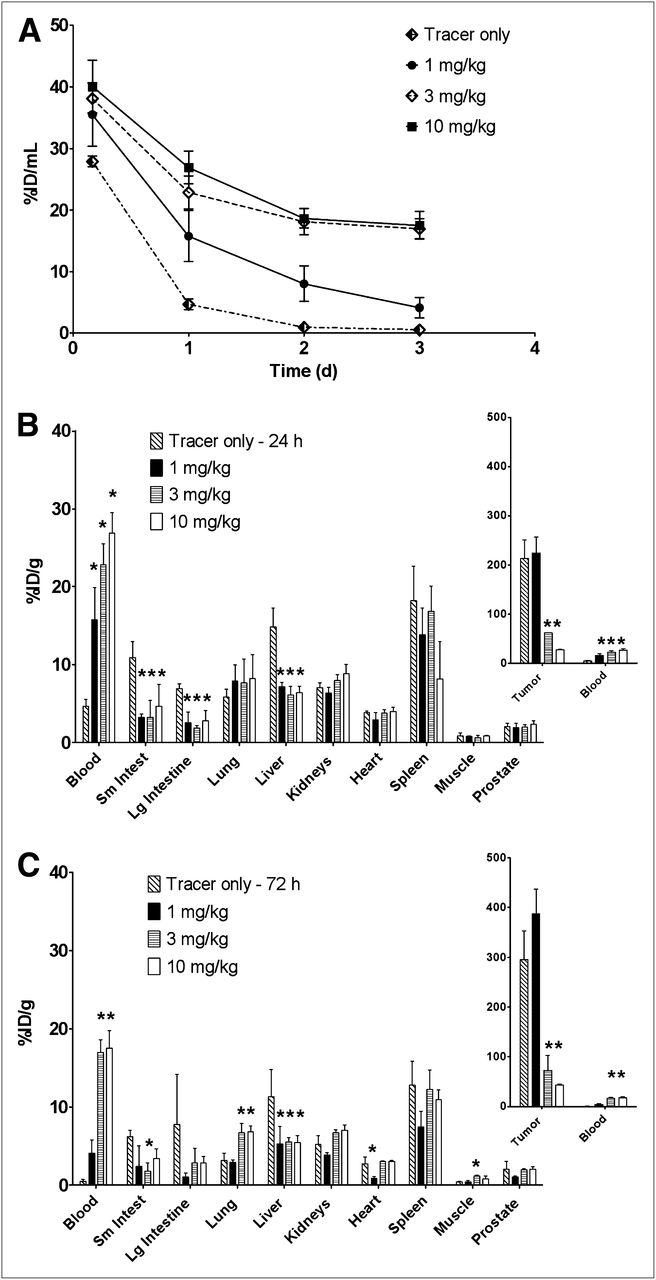
To assess whether antigen occupancy by unconjugated antibody can modulate pharmacokinetic exposure or affect the distribution of ADC between tumor and normal tissue, we predosed the tumor-bearing mice with escalating doses of anti-TENB2 antibody and monitored the uptake of 111In-anti-TENB2-MMAE in the blood, tumor, and selected tissues. A strong dose-dependent effect of predose level on blood exposure was confirmed, as reflected in blood pharmacokinetics curves, which indicated a decrease in clearance with increased predosing level (Fig. 2A). Dose-normalized exposures of 111In-anti-TENB2-MMAE, expressed as area under the concentration–time curve from 0 to 3 days (AUC0–3) values, were 23, 46, 70, and 76 percentage injected dose per milliliter (%ID/mL) × days with a 0, 1, 3 and 10 mg/kg predose, respectively. For comparison, the analogous AUC0–3 value for trastuzumab, a typical nonbinding humanized IgG1 with linear pharmacokinetics in mice, is 68 percentage injected dose per gram (%ID/g) × days (23). The rapid clearance of the cross-reactive anti-TENB2-MMAE at low doses reflects its dynamic interaction with both human and murine TENB2 in the LuCaP 77 tumor–bearing mouse model, resulting in extensive tissue and tumor uptake. The elevation of blood concentration by predose was observed as early as 4 h. For example, after 10 mg/kg predose, the blood levels of radioactivity increased from less than 30 %ID/mL (tracer only) to approximately 40 %ID/mL (Fig. 2A).

Dose-escalation studies using 111In-anti-TENB2-MMAE at 0.01 mg/kg. (A) Plasma pharmacokinetics (n = 3) of 111In-anti-TENB2-MMAE in mice that received no predose or intravenous predose of anti-TENB2 (1, 3, or 10 mg/kg) at 24 h before radiotracer administration. (B and C) Tissue distribution (n = 3) of 111In-anti-TENB2-MMAE at 24 h (B) and 72 h (C) in mice that received no predose or intravenous predose of anti-TENB2 at 24 h before radiotracer administration. *P < 0.05 vs. tracer by 1-way ANOVA.
Tumor accretion was preserved with a predose of 1 mg/kg, whereas approximately 3- and 10-fold drops in tumor uptake were observed as the predose was increased to 3 and 10 mg/kg, respectively. Specifically, mean tumor uptake (±SD) at 24 h was 214 ± 37, 225 ± 32, 62 ± 0.4, and 27 ± 0.6 %ID/g in mice receiving pretreatment of 0, 1, 3, and 10 mg of anti-TENB2 per kilogram, respectively (Fig. 2B). In contrast, liver and gut uptake of radiolabeled ADC sharply dropped after a 1 mg/kg predose at 24 h, suggesting that predosing selectively blocks ADC uptake in antigen-expressing nontumor tissues. Small intestine levels were 11 ± 2, 3 ± 0.4, 3 ± 2, and 5 ± 3 %ID/g, respectively. Hepatic levels were 15 ± 2, 7 ± 0.6, 6 ± 1, and 6 ± 0.8 %ID/g, respectively. Meanwhile, muscular uptake was low (0.6–0.9 % ID/g) and unaffected by predosing. Compared with 24-h data, the 72-h data showed overall similar trends but lower blood concentrations and higher tumor concentrations at intermediate predose levels (Fig. 2C). The high level of tumor uptake (>200 %ID/g) may be rationalized by the use of a residualizing probe, tumors that are significantly smaller than 1 g, and the high TENB2 expression level (3+) in the LuCap 77 model. Although the level of uptake (%ID/g) in the intestines is much lower than in the tumor, the gut’s contribution to blood pharmacokinetics is important because the weight of murine intestine is roughly 10-fold that of the tumors. Furthermore, the role of intestine as a clearance organ likely reduces its level of tracer residualization, thus possibly underestimating the uptake of ADC in intestines relative to that of the tumor. Also, hepatic uptake may reflect ADC binding to TENB2 antigen that has shed from the tumor (24).
We also measured the vascular and interstitial volumes of tumors relative to that of normal tissues to mechanistically understand the differential impact of predosing on uptake in tumors and in tissues not expressing tumor antigen (23,25). No significant difference in vascular volume was determined between the tumor and either small or large intestines (ranging from 8 to 13 μL of blood per gram, or about 1% blood by volume) (Supplemental Table 1; supplemental materials are available online only at http://jnm.snmjournals.org). However, the mean interstitial volume of the tumor (277 ± 47 μL of interstitial fluid per gram) was higher than that of intestine (150–157 μL/g). Taken together, these data indicate that, on extravasation, the anti-TENB2 antibody must traverse through a larger (roughly 2-fold) volume of interstitial fluid in tumor than in the gut. Although this may partially explain the preferential blockade of gut antigen over tumor antigen at 1 mg/kg, the differential expression level likely also plays a major role.
SPECT/CT
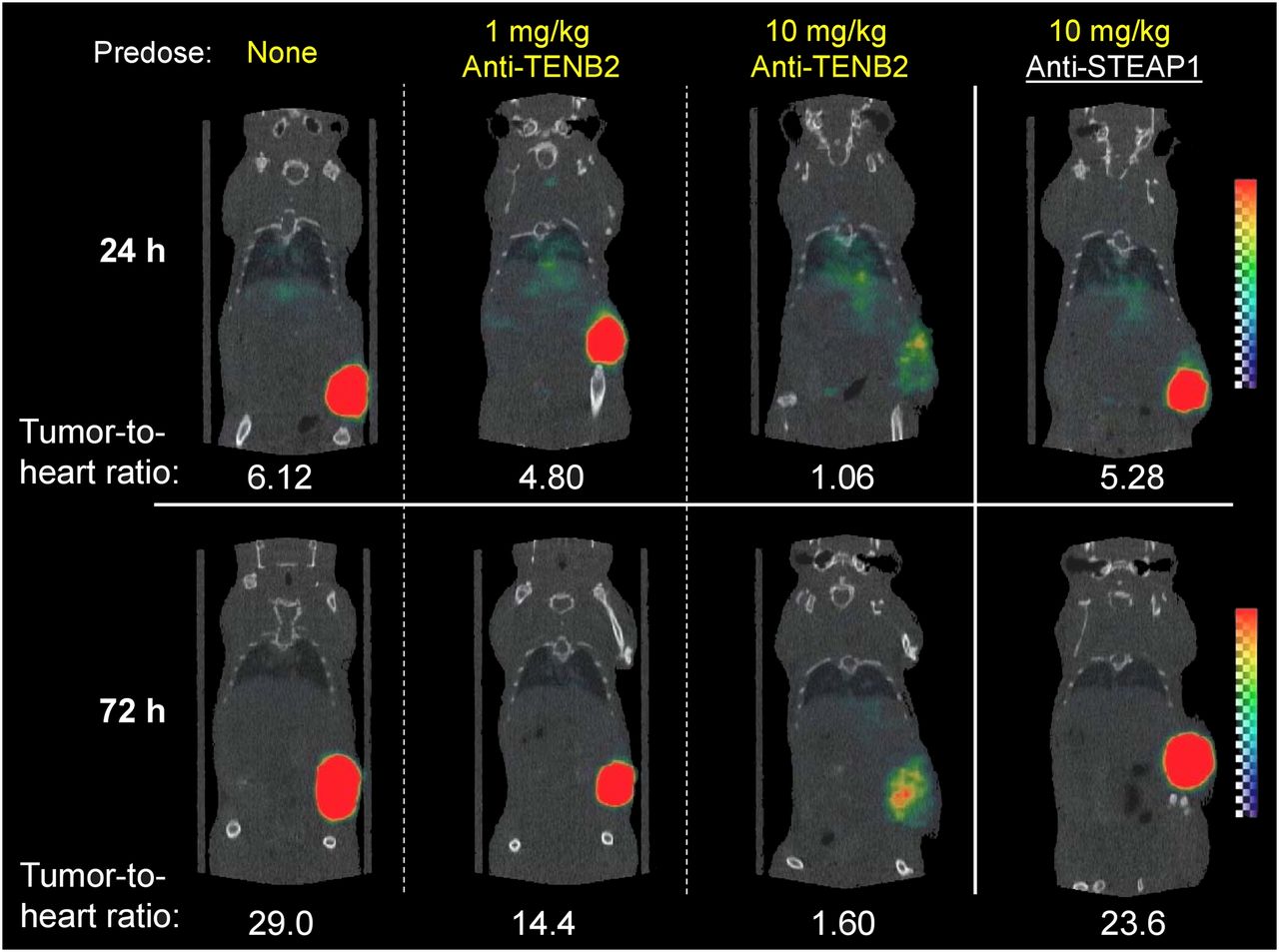
Small-animal imaging studies were conducted to further demonstrate that tumor uptake of ADC is affected by pretreatment with anti-TENB2 in an antigen-specific and dose-dependent manner. Consistent with tissue-distribution studies, pretreatment with 10 mg of anti-TENB2 per kilogram significantly reduced the tumor uptake of 111In-anti-TENB2-MMAE visible by SPECT/CT at 72 h after injection (Fig. 3). Importantly, an analogous 10 mg/kg predose of an isotype-matched control anti-STEAP1 antibody did not block tracer uptake, confirming antigen specificity of 111In-anti-TENB2-MMAE uptake in tumors. Furthermore, consistent with the dose-escalation study (Fig. 2), imaging confirmed that tracer accumulation in the tumor was not drastically reduced by a 1 mg/kg predose of anti-TENB2 on a qualitative basis.
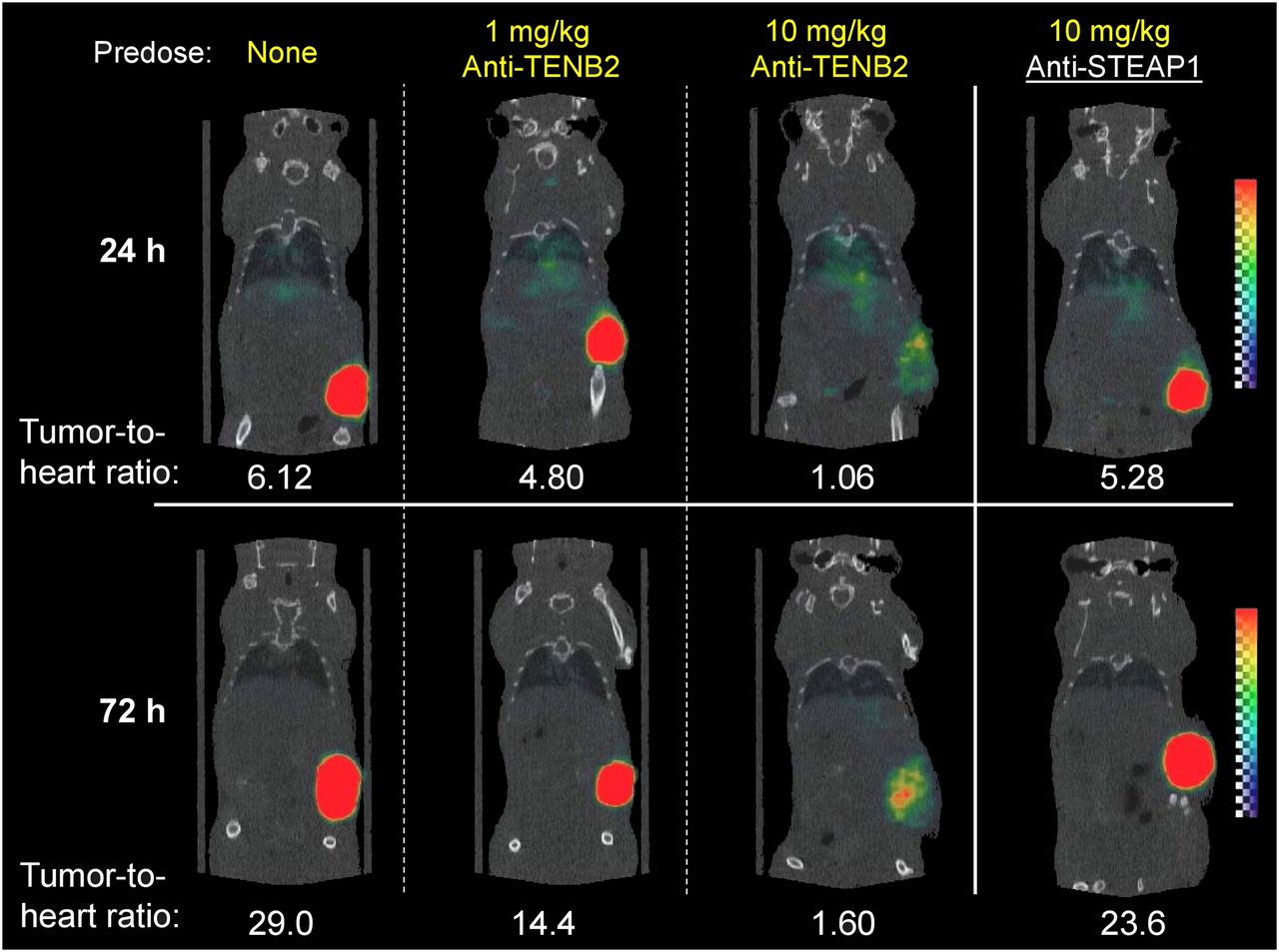
Reconstructed 3-dimensional volume-rendered SPECT/CT fusion images of 111In-anti-TENB2-MMAE (3 mg/kg) at 24 h or 72 h after injection. At 24 h before administration of radiotracer, mice received doses as indicated. All biologics were administered intravenously.
Image quantitation showed a gradual increase in tumor-to-heart ratios from 1 to 3 d with increases from 6 to 29, 5 to 14, 1 to 1.6, and 5 to 24 when pretreating with 0, 1, and 10 mg of anti-TENB2 per kilogram and 10 mg of anti-STEAP1 per kilogram, respectively (Fig. 3). These results were corroborated by γ-counting of harvested tissues after imaging, with terminal (3-d) tumor-to-blood ratios of 20, 8, 2, and 19, respectively (data not shown). Image analysis did indicate a noticeable decrease in the tumor-to-blood ratio even at the 1 mg/kg predose level. This finding was likely due to the slightly higher blood exposure after predose.
Efficacy Study
To further evaluate the impact of ADC uptake on tumor-growth inhibition, we compared the efficacy of anti-TENB2-MMAE in an TENB2-expressing LuCaP 77 model with and without predosing with unconjugated antibody. On the basis of the results from the dose-escalation study, a predose level of 0.75 mg/kg was selected to maximize the tumor uptake, and 2 dose levels of anti-TENB2-MMAE (0.75 and 2.5 mg/kg) were selected to explore the dose dependency of the potential impact from predosing. Comparable tumor inhibitions were observed at both a 0.75 and a 2.5 mg/kg dose of anti-TENB2-MMAE with or without predosing at 0.75 mg/kg (Fig. 4A). This comparability indicated that the predose of anti-TENB2 before anti-TENB2-MMAE did not compromise the overall efficacy.

In vivo LuCaP 77 tumor inhibition with anti-TENB2-MMAE. (A) anti-TENB2-MMAE was administered at 0.75 (red) or 2.5 (blue) mg/kg intravenously in single dose to male SCID beige mice bearing established LuCaP 77 tumors. Predose of anti-TENB2 (0.75 mg/kg) was administered to selected groups (open symbols and dashed lines) at 24 h before ADC. In comparison with vehicle control (black), anti-TENB2-MMAE effectively inhibited LuCaP 77 tumor growth. Mean (±SD) tumor volume of all groups vs. time is depicted. (B) Concentration–time profiles from sparse plasma sampling of anti-TENB2-MMAE at 0.75 (red) and 2.5 (blue) mg/kg in presence (open symbols and dashed lines) and absence (closed symbols and solid lines) of 0.75 mg/kg predose of anti-TENB2.
To explore the relationship between pharmacodyamic endpoint and pharmacokinetic exposure, we also compared the systemic concentrations of ADC between the groups. Consistent with the dose-escalation study, exposure of anti-TENB2-MMAE at 0.75 mg/kg was significantly boosted by a predose of anti-TENB2 antibody at 0.75 mg/kg (Fig. 4B). In contrast, the impact of a 0.75 mg/kg predose on exposure was diminished at the higher dose level of 2.5 mg/kg, suggesting that antigen-mediated clearance was largely saturated at this dose. The apparent lack of translation from higher exposure to improved efficacy at the 0.75 mg/kg ADC dose level may reflect a counterbalance between receptor occupancy and systemic exposure in the overall efficacy. Furthermore, the LuCap 77 model is a transplant model derived from a hormone-refractory prostate cancer patient whose resistance to chemotherapy may also contribute to the narrow dose response.
DISCUSSION
The development of novel ADC therapies represents a promising strategy in the treatment of prostate cancer (26,27). However, target expression in normal tissue poses special challenges for ADC development. To demonstrate proof of concept, we tested the hypothesis that predosing of unconjugated anti-TENB2 at an optimal dose will saturate specific binding sites for the antibody in normal tissue sinks while retaining sufficient tumor uptake. This approach relies on a similar biodistribution, but different toxicity profiles, between antibody and ADC. In previous studies using the same methodology, we observed only slight differences in clearance and tissue distribution between an ADC and its corresponding naked antibody (19); these differences may be due to altered molecular hydrophobicity or isoelectric point. Larger changes in tissue uptake may occur with a higher ratio of conjugation, leading to larger shifts in isoelectric point (Δ pI > 1) (28).
Predosing with nonradioactive antibodies has been successfully applied in radioimmunotherapy to increase tumor targeting of radiolabeled antibodies by blocking nonmalignant binding sites such as circulating and splenic B cells (29). For example, predosing is used in approved radioimmunotherapy approaches using both 90Y-ibritumomab tiuxetan (Zevalin; Bayer Schering Pharma AG) and 131I-tositumomab (Bexxar; GlaxoSmithKline) and has become standard practice in radioimmunotherapy targeting the CD20 antigen (30,31). However, few examples exist for translation of such dosing strategies for ADCs (32). Predosing has been explored in the clinical imaging of ovarian carcinomas (33) and was also shown to decrease spleen uptake in imaging of non-Hodgkin lymphoma patients (34). Predosing of anti-CD20 antibodies has been demonstrated to act in an antigen-specific manner (35) and improve exposure of radioimmunotherapy agents (36); however, the success of this approach is expected to depend on the predose level (37). In addition to predosing, other methods for enhancing tumor uptake of antibody therapeutics have been reported, including pretreatment with a vasoconjugate (38).
For both ADCs and radioimmunotherapy agents, the therapeutic strategy involves selective delivery of a cytotoxin (drug or radionuclide) to tumors via the antibody. However, important distinctions exist between these 2 therapeutic modalities. For example, ADCs require internalization into the endosomes or lysosomes for efficacy, whereas radioimmunotherapy agents are able to emit radiation, even from the cell surface, to achieve cell killing after direct binding to membrane antigens. Furthermore, radioimmunotherapy can deliver high levels of radiation even with low doses of radioimmunoconjugate, compared with ADCs.
Importantly, most clinically successful radioimmunotherapy agents to date have been against hematologic tumors (39), whereas our current strategy involves delivery of ADC to a solid tumor. Various impediments to the delivery of antibodies to solid tumors have been widely discussed and studied, especially in the context of microspatial distribution (40). Accordingly, it is plausible that a predose may have a differential ability to block low-level antigen expression in a highly perfused tissue sink, while leaving most antigens in a less-readily accessible solid tumor microenvironment available for subsequent ADC therapy.
Escalation of the anti-TENB2 predose had a differential, dose-dependent effect on 111In-anti-TENB2-MMAE uptake in tumor versus normal tissues (Fig. 2). These data highlighted the presence of saturable antigen-mediated uptake in both tumor and selected peripheral tissues and established the sensitivity of tumor uptake to predose levels. Furthermore, the findings confirmed our hypothesis that predosing may saturate the nontumor antigen sink at low doses, and this effect may be due to lower expression levels or greater accessibility of TENB2 in nonmalignant tissues relative to the tumor. Characteristics of the interstitial fluid space may also play a role, because a 2-fold-higher interstitial volume for the LuCaP 77 explants relative to gut (Supplemental Table 1) would require that the anti-TENB2 antibody diffuse through longer distances from the point of extravasation to the cell surface antigen. The interstitial volume is inversely proportional to the effective concentration of antibody in the biophase (i.e., the fluid in direct contact with cell surface TENB2), which is a critical determining factor in both receptor occupancy and efficacy. Conceivably, ADC uptake in the tumor could be affected differently from uptake in tissues not expressing tumor antigen, thereby presenting an opportunity to extend the therapeutic window with deliberate selection of predose levels or time intervals.
Consistent with tissue-harvest studies, tumor uptake by SPECT/CT (Fig. 3) was compromised by pretreatment with 10 mg of anti-TENB2 antibody per kilogram. In contrast, tumor uptake was largely preserved after a predose of anti-TENB2 (1 mg/kg) or control IgG anti-STEAP1 (10 mg/kg), demonstrating both dose dependency and antigen specificity. Anti-STEAP1 was selected as a negative control for its comparable target expression in the LuCaP 77 model, and the results indicated that the uptake of another antibody in the same tumor minimally affected specific interactions between TENB2 antibody and anti-TENB2-MMAE. The low levels of radioactivity in nonmalignant tissues, residualization of 111In within tumors, and observed specific blocking of 111In uptake by competitive inhibition are all consistent with successful delivery of MMAE to TENB2-expressing tumor cells.
To evaluate the impact of predosing and enhanced tumor uptake on efficacy, and to probe the delicate balance between blocking tissue uptake versus displacing anti-TENB2-MMAE at the tumor site, we compared the tumor-growth inhibition with or without predosing. Anti-TENB2-MMAE administration with predosing preserved efficacy at 2 dose levels (Fig. 4A). A predose level of 0.75 mg/kg of unconjugated antibody was selected on the basis of the ability of a 1 mg/kg predose to preserve tumor uptake in earlier studies, whereas 0.75 and 2.5mg/kg doses of anti-TENB2-MMAE were selected on the basis of their respective approximations to the inhibitory concentration of 50% and the inhibitory concentration of 90%. As expected for molecules with significant target-mediated clearance, ADC exposure at the 0.75 mg/kg level was significantly enhanced by predosing, whereas a minimal difference was observed at the 2.5 mg/kg level (Fig. 4B). The apparent disconnect between higher systemic exposure of ADC and similar efficacy at 0.75 mg/kg may be attributed to the sensitivity of tumor response to similar maximum initial dose levels with and without predosing, rather than sustained exposure over time. In addition, displaced antigen sites from predosing may also offset the potential gain. Our study did not directly evaluate the impact of predosing on the toxicity of ADC uptake in normal tissues. However, on the basis of the rapid saturation of nontumor antigen sites by inert unconjugated antibody and drastically diminished ADC uptake, it is reasonable to postulate that a more favorable safety profile, and hence improved therapeutic window, may result from the application of the predosing approach to ADC therapy. Overall, the risk of toxicity is likely reduced without affecting efficacy, which, in turn, widened the therapeutic window of our ADC.
CONCLUSION
This work has demonstrated considerable value in using preclinical studies to understand the relationships among predose level, nontumor distribution, and tumor uptake of a targeted therapeutic. Furthermore, these findings serve as a proof of principle in dosing strategies for ADCs with off-target uptake. Our work provides evidence that efficacy in ADC therapy can be maintained even after a predose of unconjugated antibody to block undesirable uptake in nonmalignant tissues. Predosing approaches could provide a viable alternative to conventional dosing strategies and hold the potential for improved safety profiles for ADC regimens.
DISCLOSURE STATEMENT
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank Michelle Schweiger, Bernadette Johnstone, Cynthia Young, Elizabeth Torres, Kirsten Messick, Jason Ho, Jose Imperio, Maria Duran, Mayra Monett, Nina Ljumanovic, Roxanne Vega, Nicole Valle, and Sheila Ulufatu for animal studies support. We also thank Daniela Bumbaca and Paul J. Fielder for helpful discussions. All authors are employees of Genentech, a member of the Roche Group, and hold financial interest in Hoffmann-La Roche. All financial support was provided by Genentech. No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Jul. 7, 2012.
- © 2012 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication January 13, 2012.
- Accepted for publication April 30, 2012.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Bioanalytical Assays for Pharmacokinetic and Biodistribution Study of Antibody-Drug Conjugates
- New Technologies Bloom Together for Bettering Cancer Drug Conjugates
- Antibody Co-Administration Can Improve Systemic and Local Distribution of Antibody-Drug Conjugates to Increase In Vivo Efficacy
- Genetic signature of prostate cancer mouse models resistant to optimized hK2 targeted {alpha}-particle therapy
- Blocking of Glucagonlike Peptide-1 Receptors in the Exocrine Pancreas Improves Specificity for {beta}-Cells in a Mouse Model of Type 1 Diabetes
- Genetic signature of prostate cancer resistant to optimized hK2 targeted alpha-particle therapy
- Molecular Imaging of Immunotherapy Targets in Cancer
- An Effective Immuno-PET Imaging Method to Monitor CD8-Dependent Responses to Immunotherapy
- Absorption, Distribution, Metabolism, and Excretion Considerations for the Development of Antibody-Drug Conjugates
- Engineered antibody fragments for immuno-PET imaging of endogenous CD8+ T cells in vivo