


Abstract
Arachidonic acid (AA) is found in high concentrations in brain phospholipids and is released as a second messenger during neurotransmission and much more so during neuroinflammation and excitotoxicity. Upregulated brain AA metabolism associated with neuroinflammation has been imaged in rodents using [1-14C]AA and with PET in Alzheimer disease patients using [1-11C]AA. Radiotracer brain AA uptake is independent of cerebral blood flow, making it an ideal tracer despite altered brain functional activity. However, the 20.4-min radioactive half-life of 11C-AA and challenges of routinely synthesizing 11C fatty acids limit their translational utility as PET biomarkers. Methods: As a first step to develop a clinically useful 18F-fluoroarachidonic acid (18F-FAA) with a long radioactive half-life of 109.8 min, we report here a high-yield stereoselective synthetic method of nonradioactive 20-19F-FAA. We tested its in vivo pharmacokinetics by infusing purified nonradioactive 19F-FAA intravenously for 5 min at 2 doses in unanesthetized mice and measured its plasma and brain distribution using gas chromatography–mass spectrometry. Results: Incorporation coefficients of injected 19F-FAA into brain phospholipids (ratio of brain 19F-FAA concentration to plasma input function) were 3- to 29-fold higher for choline glycerophospholipid and phosphatidylinositol than for ethanolamine glycerophospholipid and phosphatidylserine at each of the 2 tested doses. The selectivities and values of incorporation coefficients were comparable to those reported after [1-14C]AA (the natural arachidonate) infusion in mice. Conclusion: These results suggest that it would be worthwhile to translate our stereoselective synthetic method for 19F-FAA to synthesize positron-emitting 18F-FAA for human brain AA metabolism in neuroinflammatory disorders such as Alzheimer disease.
- positron emitting tomography (PET)
- fluorinated arachidonic acid
- fluoroarachidonic acid
- brain
- phospholipids
- arachidonic acid signaling
- imaging
- neuroinflammation
- excitotoxicity
- biomarker
- Alzheimer’s disease
- cancer
Arachidonic acid (AA; 20:4n-6) is a polyunsaturated fatty acid found in high concentration in brain phospholipids and preferentially incorporated into phosphatidylinositol and choline glycerophospholipids (CGP) (1,2). AA release from the stereoselective sn-2–numbered position of brain phospholipids is mediated by AA-selective calcium-dependent cytosolic phospholipase A2 (group IVA), which is coupled to G-protein receptors (serotonin 5-hydroxytryptamine 2A and 2C, cholinergic muscarinic M1, 2, and 3, and dopaminergic D2) or ionotropic N-methyl-d-aspartate receptors (3). AA metabolism is upregulated in neuroinflammation, excitotoxicity, and other brain disturbances, including Alzheimer disease, bipolar disorder, stroke, and HIV-1 associated dementia (4–8).
Regional disturbances in brain AA metabolism have been imaged with [1-14C]AA in animals using ex vivo autoradiography—which shows upregulated AA metabolism during neuroinflammation and excitotoxicity induced by bacterial lipopolysaccharides or N-methyl-d-aspartate (9,10) and during G-protein neuroreceptor signaling (3,11)—and in an HIV transgenic rat model of neuroinflammation (4,12). In humans, brain AA metabolism has been imaged with PET using 11C-AA as a radiotracer, at rest and during functional activation (6,13,14). Measurements have been shown to be independent of changes in cerebral blood flow and thus are markers of metabolism (13,15). Upregulated 11C-AA metabolism associated with neuroinflammation has been demonstrated in Alzheimer disease patients with PET, particularly in regions reported to have high densities of senile plaques and activated microglia (6), suggesting the utility of 11C-AA for imaging regional disturbances in brain AA metabolism in neurodegenerative disorders with a neuroinflammatory component.
Because of the short radioactive half-life (20.4 min) of 11C-AA, its routine use as a biotracer currently is limited to a few centers able to synthesize it onsite and to administer it directly to patients. Also, challenges with the synthetic chemistry caused by using an unstable bromo fatty acid precursor (2) in a Grignard reaction, and the inability to capture high-resolution images because of the short half-life of the 11C fatty acid tracer, have limited the exploitation of this tracer as an in vivo biomarker of disturbed brain AA metabolism (16,17). Thus, gaining wider clinical use of 11C-AA as a biomarker for neuroinflammation, particularly in clinical centers and hospitals that cannot synthesize it, would require the development of a stable analog with a longer radioactive half-life.
Because of the longer radioactive half-life of 18F-fluorinated compounds (109.8 min) relative to 11C tracers (20.4 min) (18–22), the use of radiofluorinated tracers in lieu of 11C-labeled tracers (e.g., 18F-FDG for 11C-glucose and 3′-deoxy-3′-18F-fluorothymidine for 11C-thymidine (23)) has provided a breakthrough in the field of PET of regional brain metabolism in normal and pathologic conditions including neuroblastoma, infection, and cancer. Following the trend to cultivate a clinically useful imaging marker with fluorinated analogs for 11C, fluorinated AA (18F-FAA) was developed by other investigators (24). But high yields of the fatty acid tracer are difficult to synthesize with the published method (25). Also, incorporation and rates of incorporation of 18F-FAA into specific brain phospholipids was not confirmed in vivo, although its presence in brain total lipid and aqueous extracts was reported in mice (25). The phospholipid kinetics and distribution are keys to validating FAA as a tracer for AA.
In the present study, we developed a high-yield synthetic method for nonradioactive, fluorinated 19F-FAA and applied our in vivo kinetic infusion model (2,26) to quantify its incorporation and rates of incorporation in brain phospholipids of unanesthetized mice using gas chromatography–mass spectrometry (GC/MS). We used GC/MS and 19F-FAA for this study because we could identify and quantitate the mass of the FAA in each phospholipid in the brain with high sensitivity, whereas had we used 18F-FAA, we would have had minimal counting rates after the extraction and would have needed to rely on GC retention times with standard reference compounds to identify the FAA. The use of GC/MS and 19F-FAA is a necessary yet insufficient step, but it has the potential to validate FAA as a true tracer of AA.
On the basis of the reported distribution of [1-14C]AA in brain phospholipids in mice (2), we hypothesized that the fluorinated AA would be preferentially incorporated into brain cholineglycerophospholipid (ChoGpl) and phosphatidylinositol (Ptdlns) after a 5-min intravenous infusion. [1-14C]AA does not differ chemically from the natural compound; thus, its incorporation is a direct measure of incorporation and distribution of naturally occurring unesterified AA. Our results confirmed the preferential incorporation of 19F-FAA into ChoGpl and Ptdlns—an incorporation pattern that was similar to that reported for 14C-AA in mice and took place at an equivalent rate (2).
MATERIALS AND METHODS
Chemical Synthesis
Reagents.
AA was obtained from NU-Check Prep. All other chemicals and solvents were purchased from Alfa Aesar, Sigma-Aldrich, or Fisher Chemicals and were used without further purification. Melting points are reported uncorrected for differences in equipment and conditions. 1H nuclear magnetic resonance (NMR) spectra were recorded on a Varian 500 NMR instrument, and low-resolution mass spectral data were obtained at the Chemistry Department of the University of California, San Diego. Column chromatography was done on silica gel (Merck grade, 230–400 mesh, 6 nm [60 Å]).
Synthetic Chemistry
The synthetic chemistry followed the general reaction scheme of Nagatsugi et al. (24). The reactions before the synthesis of methyl 20-(tert-butyldimethylsilyloxy)-5,8,11,14-eicosatetraeonate (12) were not reported in their publication.
Synthesis of Tert-Butyl(6-Chlorohexyloxy)Dimethylsilane (5).
Imidazole (5.48 g, 80 mmol) and tert-butyldimethylchlorosilane (12.13 g, 80 mmol) were added sequentially at 0°C under N2 to a stirred solution of alcohol (4) (10 g, 73 mmol) in dimethylformamide (40 mL) before a catalytic amount of 4-(dimethylamino) pyridine (DMAP) (0.09 g, 7.3 mmol) was added to the reaction mixture. After being stirred for 15 h at room temperature, the reaction mixture was quenched with a saturated NH4Cl (20 mL) solution and extracted with CH2Cl2 (100 mL). The organic extract was dehydrated with Na2SO4 and concentrated in vacuo. Purification by column chromatography (SiO2, 1% EtOAc in hexane eluant) gave a pure compound (16.3 g, 89%) as clear oil (5).
Synthesis of Tert-Butyl(6-Iodohexyloxy)Dimethylsilane (6).
NaI (14.9 g, 99.6 mmol) was added at room temperature to a stirred solution of tert-butyl(6-chlorohexyloxy)dimethylsilane (5) (5 g, 19.9 mmol) in acetone (50 mL), and the reaction mixture was refluxed for 10 h. The acetone solvent was evaporated in vacuo to obtain a residue; the residue was redissolved in CH2Cl2 and washed sequentially with Na2S2O3, water, and finally a saturated brine solution. The organic extract was dried with Na2SO4, concentrated in vacuo, and purified by column chromatography (SiO2, 1% EtOAc in hexane eluant) to give a pure compound (6.5 g, 95%) as a light yellow oil (6).
Synthesis of (6-(Tert-Butyldimethylsilyloxy)Hexyl)Triphenylphosphonium Iodide (7).
A mixture of triphenylphosphine (3.8 g, 14.6 mmol) and iodo compound (6) (5 g, 14.6 mmol) in acetonitrile (50 mL) was refluxed for 10 h under an N2 atmosphere. The mixture was cooled to room temperature and concentrated in vacuo. The pure Wittig salt was obtained by the purification of the residue using column chromatography (SiO2, 3% MeOH in dichloromethane [DCM] as eluant) as a gummy substance (7) (8.2 g, 93%).
Synthesis of 14, 15-Epoxyeicosa-cis-5,8,11-Trienoic Acid (8).
1,1′-carbonyldiimidazole (2.26 g, 14.0 mmol) was added under N2 at 0°C to a stirred solution of AA (4.4 mL, 13.3 mmol) dissolved in anhydrous CH2Cl2 (60 mL). After being stirred for 40 min, the reaction mixture was added to ethereal H2O2 solution (135 mL, 560 mmol) containing a catalytic amount of lithium imidazole (16 mg) at 0°C. KHSO4 (26 g, 190 mmol) was added after the addition of half of the arachidonyl imidazolide solution, and the resulting mixture was stirred for an additional 5 min. The reaction mixture was filtered, washed with water and brine, and dried over Na2SO4 overnight. The solvent was evaporated under reduced pressure to obtain the title compound as viscous oil (8) (3.8 g), which was used for the next reaction without any further purification.
Synthesis of Methyl 14,15-Epoxyeicosa-cis-5,8,11-Trienoate (9).
Epoxy AA (3.8 g) (8) was added under N2 at 0°C to a solution of 1,1′-carbonyldiimidazole (2.5 g, 15.9 mmol) in anhydrous CH2Cl2 (50 mL). After the mixture was stirred for 30 min, MeOH (5 mL) was added and the reaction stirred for another 4 h at room temperature. The solvent was concentrated in vacuo, and the residue was redissolved in ether, washed with water and brine, and dried over Na2SO4. After the solvent was evaporated, the purification of the residue by column chromatography (SiO2, 10% EtOAc in hexane as eluant) gave the pure title compound as a viscous oil (9) (1.4 g, 55%).
Synthesis of Methyl 14, 15-Dihydroxyeicosa-cis-5,8,11-Trienoate (10).
Cold 10% HClO4 (9.1 mL) at 0°C was added to a stirred solution of epoxy ester (1.1 g, 3.2 mmol) in tetrahydrofuran (20 mL). The reaction mixture was stirred for another 4 h, followed by quenching with aqueous NaHCO3, and then was extracted with ethyl acetate, washed with water and brine, dried over Na2SO4, filtered, and concentrated in vacuo to obtain crude compound. Purification by column chromatography (SiO2, hexane–20% EtOAc in hexane as eluant) afforded the title compound as a viscous oil (10) (0.78 g, 68%).
Synthesis of Methyl 20-(Tert-Butyldimethylsilyloxy)-5,8,11,14-Eicosatetraeonate (12).
Compound (12) was prepared by Wittig reaction according to the modified procedure of Nagatsugi et al. (24). The crude compound was purified by column chromatography (SiO2, hexane–2% EtOAc in hexane as eluant) and afforded the pure title compound as a colorless oil (12). 1H NMR (400 MHz, CDCl3) analysis provided the following results: δ 0.05 (s, 6H), 0.89 (s, 9H), 1.50–1.21 (m, 6H), 1.76–1.66 (m, 2H), 2.27–2.03 (m, 4H), 2.32 (t, J = 7.6 Hz, 2H), 2.86–2.78 (m, 6H), 3.60 (t, J = 6.6 Hz, 2H), 3.67 (s, 3H), 5.45–5.30 (m, 8H).
Synthesis of Methyl 20-Hydroxy-5,8,11,14-Eicosatetraenoate (13).
Tetrabutylammonium fluoride (0.17 mL, 0.17 mmol, 1 M tetrahydrofuran solution) was added at 0°C under an N2 atmosphere to a stirred solution of tert-butyldimethylsilyl–protected alcohol (12) (0.051 g, 0.11 mmol) in anhydrous tetrahydrofuran (2 mL). The reaction mixture was warmed to room temperature and stirred for 5 h. It was quenched with saturated aqueous NH4Cl solution, extracted with EtOAc, washed with brine, dried (Na2SO4), and concentrated in vacuo. Purification by column chromatography (SiO2, hexane–10% EtOAc in hexane as eluant) afforded the title compound as a clear oil (13) (0.036 g, 96%). 1H NMR (400 MHz, CDCl3) analysis revealed the following information: δ 1.27 (br, 1H), 1.60–1.36 (m, 6H), 1.68–2.15 (m, 2H), 2.33 (t, J = 7.6 Hz, 2H), 2.86–2.78 (m, 6H), 3.60 (t, J = 6.6 Hz, 2H), 3.67 (s, 3H), 5.45–5.30 (m, 8H). 13C NMR (100 MHz, CDCl3) analysis provided the following results: δ 24.6, 25.3, 25.4, 26.4, 27.0, 29.1, 29.3, 30.2, 32.5, 33.3, 51.3, 62.6, 127.6, 127.8, 128.3, 128.5, 128.7, 128.8, 130.0, 130.8.
Synthesis of Methyl 20-(p-Toluenesulfonyloxy)-5,8,11,14-Eicosatetraenoate (14).
Triethylamine (0.33 mL, 0.22 mmol) and p-toluenesulfonyl chloride (0.041 g, 0.22 mmol) were added sequentially at 0°C under an N2 atmosphere to a stirred solution of alcohol 13 (0.04 g, 0.11 mmol) in anhydrous CH2Cl2 (2 mL). The reaction mixture was warmed to room temperature and stirred for another 6 h. Ice-cold water was added, extracted with water, washed with water and saturated brine, dried (with Na2SO4), filtered, and concentrated under the reduced pressure. Purification by column chromatography (SiO2, hexane–2% EtOAc in hexane as eluant) gave the compound (49.3 mg, 93%) as a colorless oil (14). 1H NMR (400 MHz, CDCl3) analysis revealed the following information: δ 7.79 (d, J = 8.4 Hz, 2H), 7.35 (d, J = 8.4 Hz, 2H), 5.35 (m, 8H), 4.01 (t, J = 6.0 Hz, 2H), 3.65 (s, 3H), 2.81 (m, 6H), 2.31 (t, J = 7.5 Hz, 2H), 2.11 (m, 4H), 1.71 (p, J = 7.2 Hz, 2H), 1.62 (m, 2H), 1.39 (m, 4H). 13C NMR (100 MHz, CDCl3) analysis provided the following results: δ 21.5, 24.6, 25.2, 25.5, 26.4, 26.8, 27.0, 28.6, 30.2, 33.3, 51.4, 70.4, 127.7, 127.9, 127.9, 128.0, 128.1, 128.7, 128.8, 129.6, 129.7, 133.0, 144.5, 173.9.
Synthesis of Methyl 20-Fluoroarachidonate (15).
Tetrabutylammonium fluoride (0.35 mL, 0.35 mmol, 1 M tetrahydrofuran solution) was added at 0°C under an N2 atmosphere to a stirred solution of tosylate 14 (0.04 g, 0.089 mmol) in tetrahydrofuran (1 mL). The reaction mixture was stirred for 2 h, followed by the evaporation of solvent under reduced pressure. This gave a crude compound, which was redissolved in CH2Cl2, washed with water and brine, dried with Na2SO4, filtered, and concentrated in vacuo. Purification of column chromatography (SiO2, hexane–2% EtOAc in hexane as eluant) afforded the title compound as a pale yellow oil (15) (0.028 g, 95%). 1H NMR (400 MHz, CDCl3) indicated the following results: δ 5.38 (m, 8H, =CH), 4.47 (dt, J = 47.0, 6 Hz, 2H, CH2F), 3.67 (s, OCH3), 2.82 (m, 6H), 2.33 (t, J = 7.2 Hz, CH2), 2.11 (m, 4H, CH2), 1.71 (m, 4H, CH2), 1.42 (m, 4H, CH2). 13C NMR indicated the following results: δ 24.75, 24.83, 25.60, 26.53, 27.06, 29.20, 29.67, 30.20, 30.39, 30.2, 30.4, 33.4, 51.44, 83.25, 84.88, 127.97, 128.15, 128.19, 128.44, 128.76, 129.0, 129.95, 174.02.
Synthesis of 20-Fluoroarachidonic Acid (20-FAA) (3).
LiOH was added at 0°C under an N2 atmosphere to a stirred solution of ester (15) (0.025 g, 0.074 mmol) in tetrahydrofuran:water (5:1). The resulting mixture was stirred for 24 h at room temperature. The mixture was quenched with 1 M aqueous oxalic acid, and the solvent was removed under reduced pressure, extracted with CH2Cl2, washed with water and brine, dried over Na2SO4, filtered, and evaporated under reduced pressure to obtain a crude acid. Purification by column chromatography (SiO2, hexane–20% EtOAc in hexane as eluant) gave pure acid as a colorless oil (3) (0.22 g, 95%). 1H NMR (400 MHz, CDCl3) analysis indicated the following: δ 5.39 (m, 8H, =CH), 4.45 (dt, J = 47.2 Hz, 6 Hz, 2H, CH2F), 2.82 (m, 6H), 2.38 (t, J = 7.6 Hz, CH2), 2.12 (m, 4H), 1.73 (m, 4H, CH2), 1.42 (m, 4H, CH2). 13C NMR (100 MHz, CDCl3) analysis indicated the following: δ 24.9, 24.8, 24.8, 25.6, 26.4, 27.0, 29.2, 29.7, 30.2, 30.2, 30.2, 30.4, 33.3, 83.2, 84.9, 127.97, 128.15, 128.19, 128.44, 128.76, 129.0, 129.95, 179.52; electrospray ionization mass spectrometry: m/z 321.46(M-H+).
Pharmacokinetics in Unanesthetized Mice
Animals.
The plasma and brain kinetics of the purified 19F-FAA were tested in unanesthetized adult C57BL/6J male mice. The study was conducted following guidelines from the National Institutes of Health for the care and use of laboratory animals (27) and was approved by the Animal Care and Use Committee of the Eunice Kennedy Shriver National Institute of Child Health and Human Development. Male C57BL/6J mice were obtained at the age of 2–3 mo. The mice were maintained in an animal facility in which temperature, humidity, and light cycle were regulated with free access to water and a fixed diet (Rodent NIH-07) that contained (as fractional percentage of total fatty acids) 30.6% saturated fat, 22.5% monounsaturated fat, 47.1% linoleic acid, 4.9% α-linolenic acid, 0.2% AA, 1.6% eicosapentaenoic acid, and 2.2% docosahexaenoic acid.
Surgical Procedures and Tracer Infusion.
At 4–5 mo of age, a mouse was anesthetized with 1%–3% isoflurane, and polyethylene catheters were inserted into a femoral artery and vein. The mouse was allowed to recover from surgery in an environment maintained at 25°C, with its hindquarters loosely wrapped and taped to a wooden block. During recovery, body temperature was maintained at 37°C by means of a rectal probe and a heating element (Indicating Temperature Controller; Yellow Springs Instrument). After 3 h of recovery, the unanesthetized mouse was infused intravenously for 5 min with 130 μL of N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) buffer (pH 7.4) containing fatty acid–free bovine serum albumin (50 mg/mL; Sigma) or 0.1 (0.3 nmol) or 0.5 mg (1.6 nmol) per mouse of 20-FAA dissolved in N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid)–bovine serum albumin at a rate of 0.0223 (1 + e –0.32t) mL/min (t in seconds), using a computer-controlled variable-rate infusion pump (no. 22; Harvard Apparatus). During infusion, timed arterial blood samples (∼20 μL) were collected in polyethylene Beckman centrifuge tubes coated with heparin, lithium, and fluoride at 0, 0.25, 0.5, 1.0, 1.5, 3.0, and 4.0 min. In addition, a 150-μL sample was collected at 4.9 min. Plasma samples were separated by centrifuging at 13,000 rpm for 1 min and stored at −80°C until they were analyzed. At 5 min, the mouse was anesthetized with sodium pentobarbital (50 mg/kg, intravenously) and underwent head-focused microwave irradiation (5.5 kW, 0.9 s, 75% power output; Cober Electronics) to stop brain fatty acid metabolism (28). The brain was excised, dissected sagittally, and stored at −80°C until analysis.
Plasma and Brain Lipid Extraction and Separation.
Total lipids were extracted from plasma (7–10 μL) and from half brain (∼0.2 g) according to the method of Folch et al. (29). Heptadecanoic acid (17:0) was added as an internal standard to plasma before extraction. Total lipid extracts were separated by thin-layer chromatography on silica gel plates (Silica Gel 60A; Whatman) prewashed with chloroform–methanol (2:1 v/v) and activated by heating at 100°C. Neutral lipid subclasses—including phospholipids, unesterified fatty acids (UFAs), triglycerides, and cholesteryl esters (CEs)—were separated using a mixture of heptane:diethyl ether:glacial acetic acid (60:40:3 by volume) (30), and authentic phospholipid, cholesterol, UFA, triglyceride, and CE standards were run in separate lanes to identify the bands. Phospholipid classes (ethanolamine glycerophospholipid [EtnGpl], ChoGpl, Ptdlns, phosphatidylserine [PtdSer]] were separated in chloroform:methanol:H2O:glacial acetic acid (60:50:4:1 by volume) (31) and identified with authentic standards in separate lanes. The plates were sprayed with 0.03% (w/v) 6-p-toluidine-2-naphthalene sulfonic acid (Acros) in 50 mM Tris-HCl buffer (pH 7.4), and the lipid bands were visualized under ultraviolet light. Each band was scraped into tubes containing di-17:0 PC for quantifying phospholipid, CE, triglyceride, EtnGpl, ChoGpl, Ptdlns, and PtdSer lipid fractions or 17:0 heptadecanoic acid to quantify brain UFAs. Toluene (0.2 mL) was added to each tube containing the esterified lipids. The UFAs were extracted from the scraped silica using 2:1 chloroform–methanol and 1.5 mL of 0.1 M KCl, dried and derivatized as described below. The remaining silica extracts containing esterified fatty acids (phospholipids, CE, triglycerides, EtnGpl, ChoGpl, Ptdlns, and PtdSer) were hydrolyzed with 1 mL of 10% methanolic KOH at 70°C for 1 h, and the resulting free fatty acids were displaced with 1 mL of HCl (37%) and 1 mL of H2O and extracted with 3 mL of hexane. The top hexane layer was transferred to new tubes after being stirred in a vortex mixer, centrifuged at 3,000 rpm (5 min), and derivatized.
Derivatization.
The hydrolyzed fatty acids (in hexane) were dried and derivatized in 100 μL of pentafluorobenzyl bromide (Sigma-Aldrich) reagent containing acetonitrile, diisopropylamine, and pentafluorobenzyl (1,000:100:10 v/v/v) by shaking for 15 min. The pentafluorobenzyl reagent was dried, and the derivatized fatty acids were dissolved in 50–100 μL of hexane and transferred to vials for GC/MS analysis.
GC/MS Analysis.
GC/MS analysis was performed using a Finnigan TRACE DSQ mass spectrometer (Thermo Electron) coupled with TRACE GC. The fatty acyl pentafluorobenzyl esters in hexane were injected onto a DB-FFAP capillary column (30 m long × 0.25 mm in internal diameter, 0.25-μm film thickness; J&W Scientific) interfaced directly into the negative chemical ionization source using methane as a reagent gas and helium as a carrier gas. The GC column oven temperature was programmed from 80°C to 185°C at 20°C per minute, then to 240°C at 10°C per minute and held for 30 min. The injector and transfer lines were maintained at 240°C and 220°C, respectively. The 17:0 internal standard and FAA were identified by select ion mode of the base peak (M-pentafluorobenzyl). The concentration of FAA was quantified by relating its peak area to the area of the internal standard and correcting for the response factor, which was determined from a calibration curve of the FAA standard. The response factor for FAA was 0.2.
Calculations.
On the basis of the intravenous infusion of 19F-FAA, incorporation coefficients into individual brain phospholipids (mL/s/g × 10−4) were calculated as follows (26):
RESULTS
Chemical Synthesis
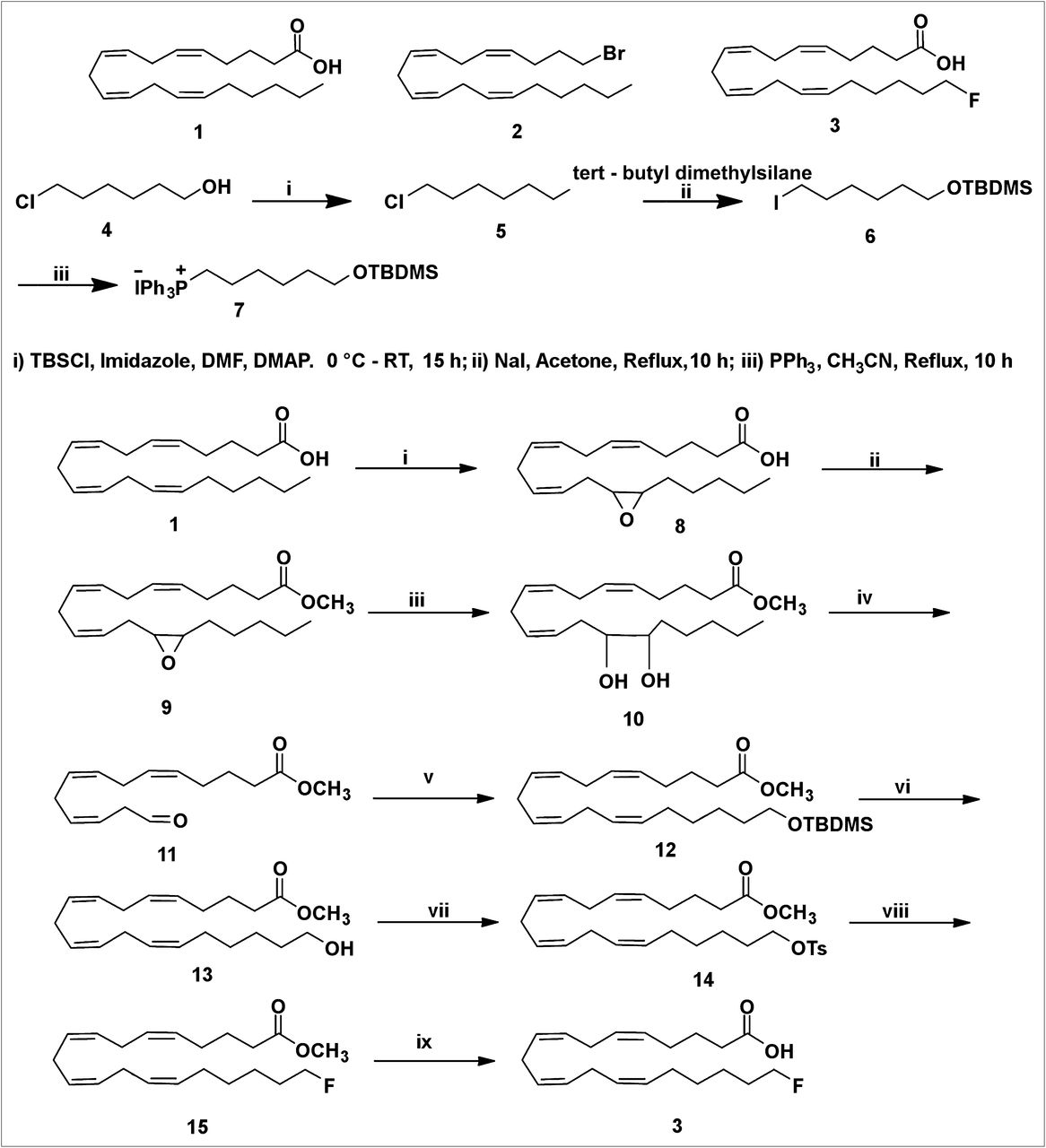
20-19F-FAA (3) was prepared from readily available AA by an improvement of the reported procedure (Scheme 1) (24). AA (1) has 4 (Z,Z) double bonds at the Δ5,6-, Δ8,9-, Δ11,12-, and Δ14,15-positions. Construction of the Z-stereochemistry in the Δ14,15-olefin of AA is essential for the preparation of 20-FAA. This investigation suggested that it may be possible to prepare FAA (3) of established geometry by cleaving the Δ14,15-olefin using reagents that do not react at the Δ5,6-, Δ8,9-, and Δ11,12-olefin sites of the AA.
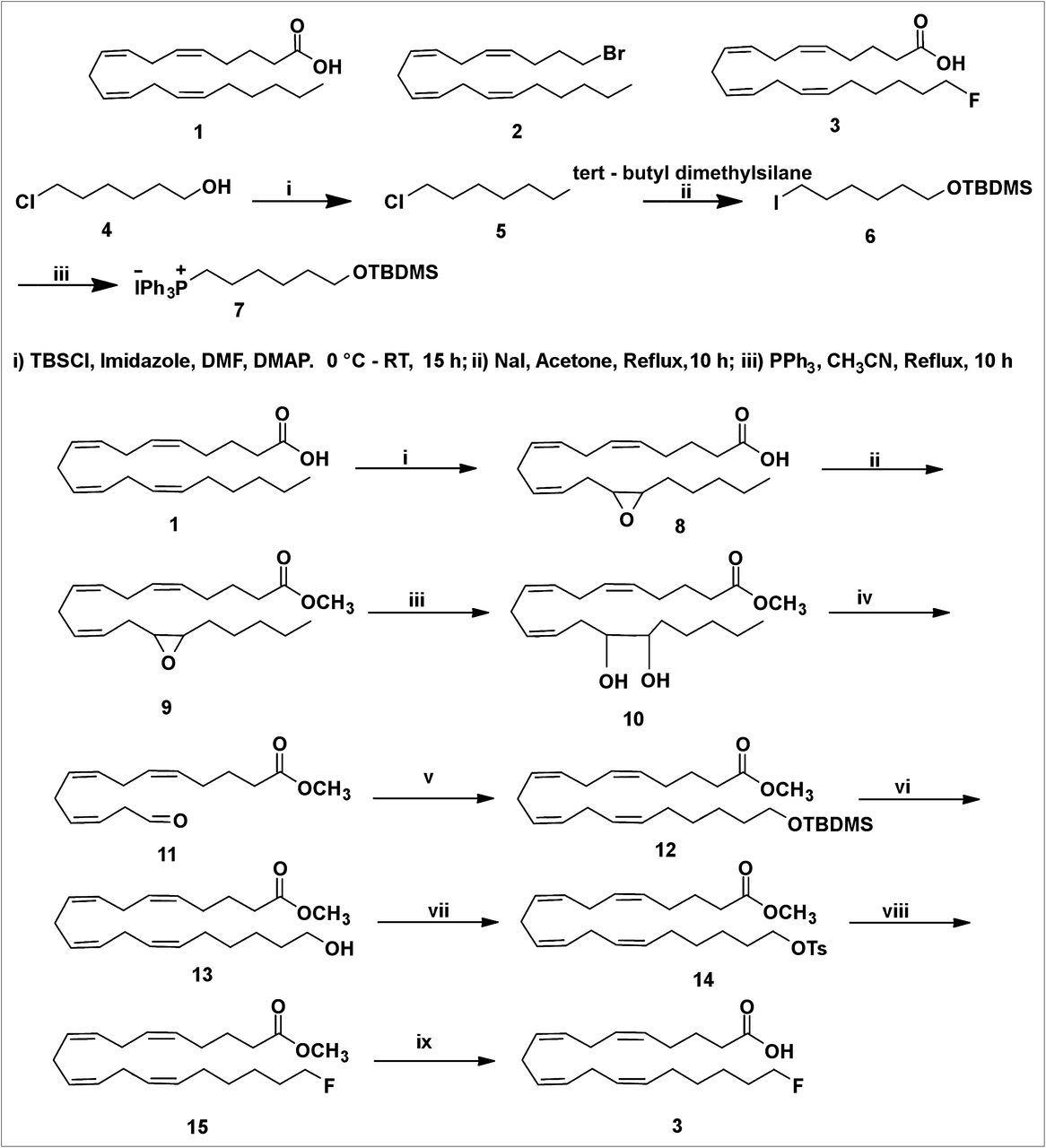
(i) 1,1′ carbonyl diimidazole, H2O2, Li imidazole, KHSO4, CH2Cl2; 0°C to room temperature, 40 min. (ii) 1,1′ carbonyl diimidazole, MeOH, CH2Cl2, 2 h, 0°C to room temperature. (iii) 10% HClO4, tetrahydrofuran, 0°C to room temperature, 4 h. (iv) Pb(OAc)4, DCM, −20°C, 30 min. (v) n-BuLi, 7 Wittig salt, tetrahydrofuran, −78°C, 3 h. (vi) Tetrabutylammonium fluoride, tetrahydrofuran, 0°C to room temperature, 3 h. (vii) TsCl, Et3N, CH2Cl2, 0°C to room temperature, 6 h. (viii) Tetrabutylammonium fluoride, tetrahydrofuran, 0°C to room temperature, 3 h. (ix) LiOH, tetrahydrofuran; H2O, room temperature, 24 h.
The m-chloroperbenzoic acid used previously is unsuitable for selective epoxidation of AA. The selective epoxidation of AA was performed by 2 different methods. First, the reaction of AA with oxalyl chloride in benzene afforded acid chloride. Oxidation of acid chloride with 50% hydrogen peroxide in the presence of pyridine and lithium hydroxide gave peroxy AA. This acid was rearranged to the epoxy acid (8). The epoxy acid was obtained in 25% yield. This method was not satisfactory for the preparation of the epoxy acid because of the low yield, poor scale up, and difficulty in handing the acid chloride.
We then investigated another method in which the AA was oxidized with a dry solution of hydrogen peroxide in diethyl ether–dichloromethane and carbonyldiimidazole in the presence of a basic catalyst, lithium imidazole, to form peroxy AA, which is selectively converted to 14,15-epoxyarachidonic acid (8) (32). This intramolecular epoxidation of peroxy AA generated the 14,15-epoxide with high efficiency. The selective formation of epoxy acid (8) from AA (1) can be ascribed to the stereoelectronic constraints for intramolecular oxygen transfer arising from attack by the C=C π-orbital electrons backside to the O–O bond (σ-orbital) of the internally hydrogen-bonded peroxycarbonyl group, with the C=C σ-plane approximately perpendicular to the peroxycarbonyl ring. The selectivity is arguably controlled by the ring strain because epoxidation requires a perpendicular orientation between C=C σ-plane and the internally hydrogen-bonded peroxy carbonyl ring. Epoxidation of the other 3 C=C double bonds—Δ5,6-olefin, Δ8,9-olefin, and Δ11,12-olefin—requires more ring strain and thus is less favored.
Of the 2 methods described here that led to the epoxy acid, the latter approach described in Scheme 1 is more versatile than the former one, which is based on the acid chloride reaction. Preparation of the epoxy ester via acid chloride and anhydride is ruled out because of the labile functional moiety, the epoxy group. There are 2 common methods to prepare the epoxy methyl ester (9). In the first method, the epoxy acid (8) was converted to the methyl ester by diazomethane treatment. This reagent was prepared in ethereal solution by the action of sodium hydroxide with N-methyl-N-nitroso-p-toluene-sulfonamide (Diazald; Aldrich Chemical Co.) in the presence of an alcohol. This procedure is difficult to scale up to gram quantities. Diazomethane is explosive and toxic. As a result, we did not continue to make epoxy methyl ester with diazomethane on a large scale. A useful alternative, which gives high yields for fatty acid components (33), is to activate the latter by reaction with 1,1′-carbonyldiimidazole to form an imidazolide, which is reacted immediately (without isolation) with methanol in the presence of a base to give the required ester (9). This procedure has been used in the preparation of epoxy methyl ester. Observation of the thin-layer chromatography of methyl ester indicated the absence of the epoxy acid (8).
The epoxide group of 9 was cleaved to a diol (10) using perchloric acid in a mixture of tetrahydrofuran and water in 55% yield. The diol (10) was cleaved with lead tetraacetate to furnish the aldehyde (11) in low yield.
A literature bromination or iodination reaction for cross coupling with this aldehyde (11) to construct the C16–C20 carbon side chain in 20-FAA was unsuccessful (34,35).
However, another approach was successful. The commercially available 6-chlorohexanol (4) was protected as its tert-butyldimethylsilyl ether. The compound (5) was obtained in 89% yield. After the protection of the hydroxyl group with the tert-butyldimethylsilyl group, the chloro compound (5) was heated to react with sodium iodide in anhydrous acetone to give an iodo compound (6) in 95% yield. The ylides themselves are generated in a 2-step sequence involving nucleophilic attack of triphenylphosphine on an iodo compound (6) to form a phosphonium salt (7). A 5-carbon extension of the labile aldehyde intermediate was performed by the Wittig reaction. This Wittig salt (7) was treated with a base n-butyllithium to generate the ylide. The Wittig salt was thus allowed to react with 2 equivalents of n-butyllithium at −78°C to ensure the formation of the ylide. The resultant ylide solution was treated with labile aldehyde (11) as shown in Scheme 1. The Wittig product was obtained as a mixture of major Z-stereochemistry in the Δ14,15-olefin and the minor E-stereochemistry Δ14,15-olefin as a low yield. The attempts to separate the 2 Δ14,15-olefin cis and trans isomers by routine column chromatography were unsuccessful.
The tert-butyldimethylchlorosilane group of the 12 was then desilylated using tetrabutylammonium fluoride to provide an alcohol (13) in 96% yield. As a result, the 2 isomers—the major Z-stereochemistry in the Δ14,15-olefin and the minor E-stereochemistry in the Δ14,15-olefin—were easily separated by flash chromatography. The stereochemistry of the isolated 13 was determined by NMR spectroscopy on the 14–15 double bonds. Only the Z isomer was detected by proton NMR spectroscopy. The alcohol was not contaminated with the trans isomer. As expected, the Δ14,15 E-alkene isomer was not detected by proton NMR spectroscopy or carbon NMR spectroscopy.
Generally, triflate precursors are unstable and thus difficult to isolate and purify. The mesylate precursor is considered to be a more stable precursor than the tosylate. But tosylation of 13 with TsCl in pyridine gave no better results. On the other hand, tosylation of 13 with TsCl in triethylamine gave the tosylate (14) in 93% yield. Nucleophilic displacement of the tosylate (14) with tetrabutylammonium fluoride in tetrahydrofuran furnished the desired fluoro compound (15) in 95% yield. Careful treatment of 15 with 3 equivalents of lithium hydroxide at 0°C to ambient temperature selectively hydrolyzed the methyl ester, which was purified by column chromatography to furnish 20-FAA (3) in 95% yield (Scheme 1). On the basis of a literature precedent, the tosylate or mesylate precursor can be used to produce 20-18F-FAA in sufficient yields, in reaction times and temperature commensurate with the half-life and stability of the precursor (36).
Pharmacokinetic Studies in Mice
The in vivo pharmacokinetics of FAA were determined in mice during a 5-min intravenous infusion of a low-FAA dose (0.3 nmol) equivalent to the 14C-AA dose used in a previous study (2) or a high dose (1.6 nmol). The FAA concentration in plasma (nmol/mL) increased over time in a dose-dependent manner (Fig. 1). The plasma area under the curve for the low and high dose was 107 ± 78 and 849 ± 68 nmol s mL−1, respectively. Statistical analysis was not performed because of the small sample sizes (n = 2–3).
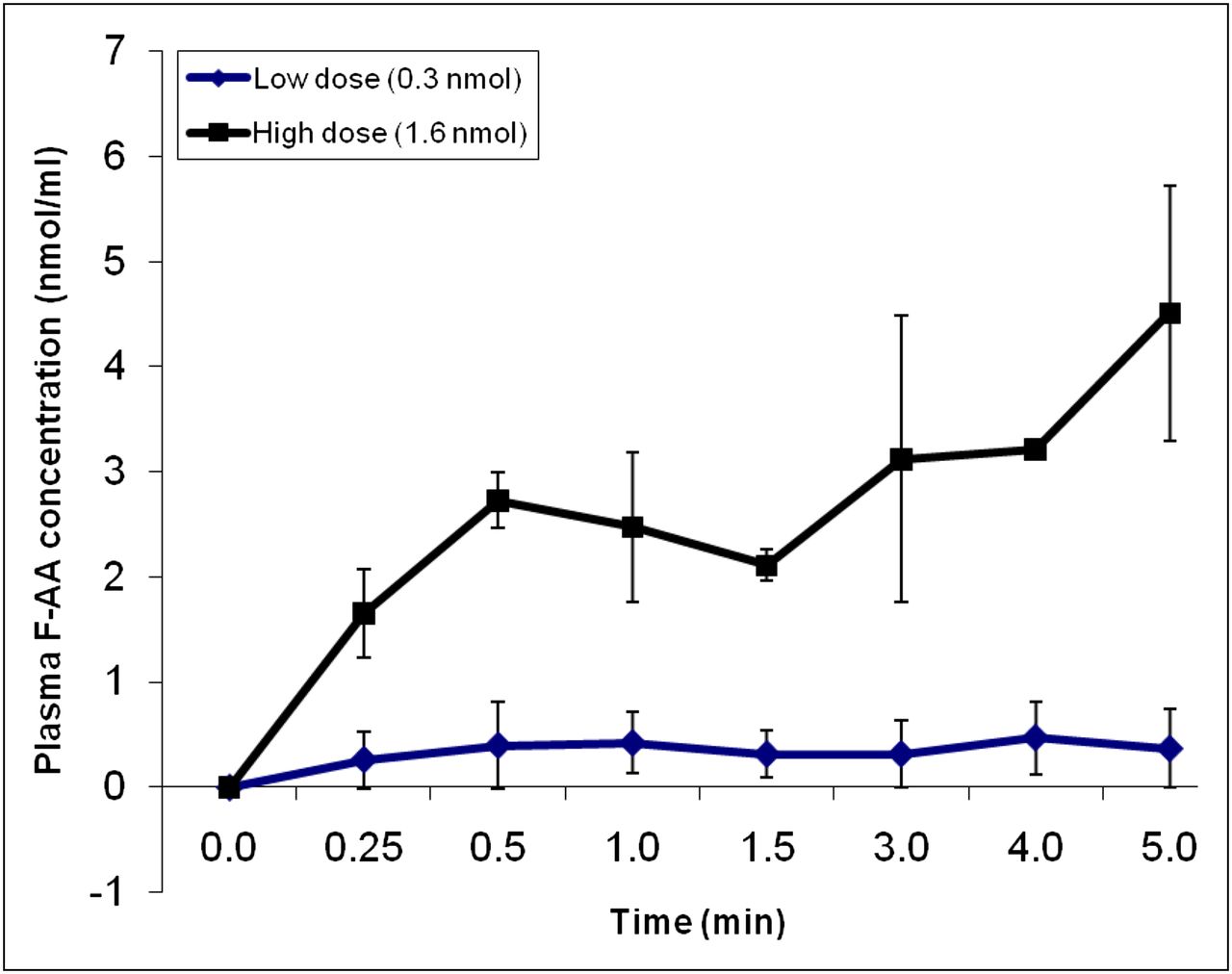
Time course of plasma FAA concentration during intravenous infusion of 0.3 or 1.6 nmol FAA over 5 min. Values are mean ± SD of n = 2 (low dose) or 3 (high dose).
DISCUSSION
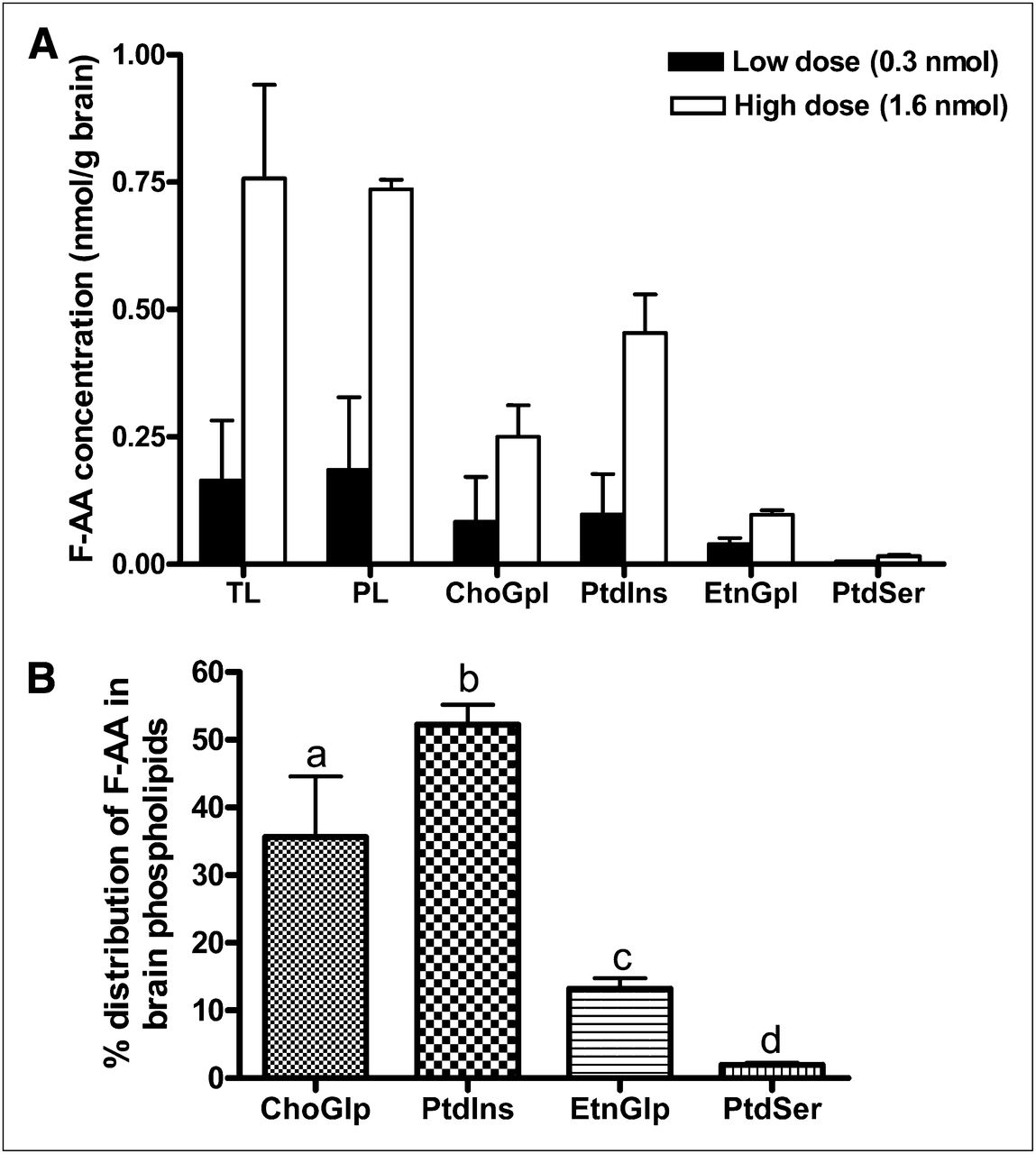
Nagatsugi et al. reported the incorporation of a 20-18F-FAA–injected brain total lipid extract but did not confirm its presence in brain phospholipids (25). In our study, GC/MS analysis revealed the presence—at the end of the 5-min infusion of 19F-FAA in brain—total lipid, total phospholipid, and individual phospholipid fractions in a dose-dependent manner (Fig. 2A). Most FAA was detected in phospholipids, and none or a negligible amount (<0.01 nmol/g) was detected in the brain UFA, triglyceride, and cholesteryl ester fractions. Within phospholipids, FAA concentration was highest in PtdIns and ChoGlp, followed by EtnGlp and PtdSer, for both doses (Fig. 2A). FAA was not detected in brain lipids of the saline and albumin–infused control mouse.

(A) FAA concentration in brain lipids (nmol/g wet weight) after 5-min infusion with low dose (0.3 nmol) or high dose (1.6 nmol). Values are mean ± SD of n = 3 mice for low dose, except for EtnGpl and PtdSer (for which n was 2 because FAA was not detected in 1 mouse), and n = 2 mice for high dose. (B) Percentage distribution of FAA in brain phospholipid subfractions. Bars represent mean ± SD. Bars with overlying letters are significantly different from each other by 1-way ANOVA, followed by Tukey post hoc test. n = 5 for ChoGpl and PtdIns, and n = 4 for EtnGpl and PtdSer because FAA was not detected in 1 mouse infused with low dose. PL = phospholipids; TL = total lipids.
Figure 2B shows the distribution of FAA in each phospholipid fraction, expressed as a percentage of the FAA concentration in total phospholipids (i.e., percentage of nanomoles of FAA in each phospholipid fraction per nanomole of FAA in total phospholipids). The mean percentages differed significantly from each other, being the highest in ChoGlp, followed by PtdIns, EtnGlp, and PtdSer (P < 0.01), suggesting preferential accumulation of FAA in phosphatidylinositol and CGP.
The incorporation coefficients, k* (Eq. 1), for both low and high doses are shown in Figure 3. As indicated, FAA was incorporated into brain total lipids and phospholipids and in individual phospholipids. k* was lower for the high-infusion dose than for the low-infusion dose, suggesting substrate inhibition at the higher dose. Within phospholipids, FAA was incorporated preferentially into PtdIns and ChoGpl at both doses (Fig. 3). This preferential incorporation is similar to the preferential incorporation of 14C-AA (1).
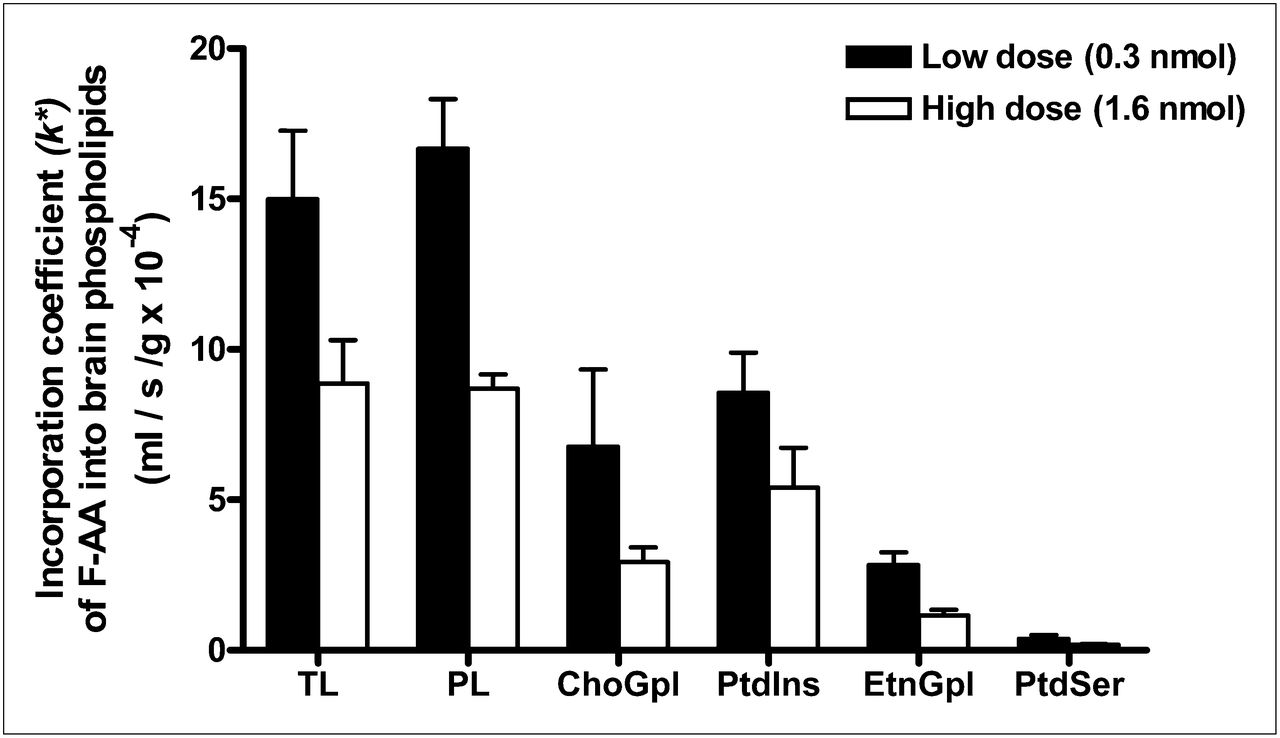
FAA incorporation coefficient, k*, in brain lipids after 5-min infusion with low dose (0.3 nmol) or high dose (1.6 nmol). Values are mean ± SD of 3 mice for low dose (black bars), except for EtnGlp and PtdSer, for which n = 2 because FAA was not detected in 1 mouse; n = 2 for high dose (white bars). PL = phospholipids; TL = total lipids.
Table 1 compares the mean values of k* for FAA in our study with published values for intravenously injected 14C-AA in mice (2). For both tracers, the values of k* into individual phospholipids are comparable and higher in PtdIns and ChoGpl than in EtnGpl and PtdSer (1,2). These results indicate that adding 19F to AA at the 20 position did not markedly change the incorporation kinetics or enzyme or phospholipid selectivities in comparison with the natural substance. Enzymes involved in incorporation are acyl-CoA synthetases, which activate AA to AA-CoA, and acyl CoA:lysophospholipid acyltransferase, which transfer AA from AA-CoA into an open sn-2 position site of a lysophospholipid (26,37,38). These enzymes show selectivities for AA, compared with other fatty acids (39,40).
Comparison of 19F-FAA k* Values to Published Values for 14C-AA
CONCLUSION
We developed a stereoselective synthesis of nonradioactive 19F-FAA yield, which was incorporated preferentially into mouse brain PtdIns and ChoGpl, as has been reported for 14C-AA, and with similar coefficients. This development should expand on present PET capabilities, allowing centers to use our method to synthesize positron-emitting 18F-FAA for PET of upregulated brain AA metabolism in clinical inflammation, which likely occurs in Alzheimer disease, bipolar disorder, schizophrenia, and Parkinson disease and also in HIV-1 associated dementia, brain trauma, epilepsy, cancer, and bacterial infections. A longer radioactive half-life than that of 11C-AA would also enhance the current resolution limitations of PET scans, because more radioactivity can be captured and integrated over time to obtain a higher-resolution image. 20-18F-FAA has the potential to be an important radiobiomarker in patient management and assessment in neurology, psychiatry, oncology, and cardiology.
DISCLOSURE STATEMENT
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We acknowledge the NCI editorial board for reviewing the paper and providing valuable comments. Financial support for this research was provided by the Society of Nuclear Medicine Covidien Seed Grant; by the Intramural Research Program of the National Institute on Aging, National Institutes of Health; and by the Intramural Program of the National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health. No other potential conflict of interest relevant to this article was reported.
Footnotes
↵* Contributed equally to this work.
Published online Jul. 31, 2012.
- © 2012 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication March 7, 2012.
- Accepted for publication April 30, 2012.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.