


Abstract
PET brain imaging of the serotonin 1A (5-hydroxytryptamine 1A [5-HT1A]) receptor has been widely used in clinical studies. Currently, only a few well-validated radiolabeled antagonist tracers are available for in vivo imaging of this central receptor. 5-HT1A receptors exist in high- and low-affinity states, depending on their coupling to G proteins. Agonists bind preferentially to receptors in the high-affinity state and thereby could provide a measure of functional 5-HT1A receptors. Therefore, it is of great interest to develop an 18F-labeled full agonist 5-HT1A receptor radiotracer. In this study, we radiolabeled the high-affinity 5-HT1A receptor agonist 18F-F13714 and investigated its potential as a PET tracer. Methods: F13714 nitro precursor was synthesized and radiolabeled via a fluoronucleophilic substitution. In vitro binding assays were performed using established protocols. Radiopharmacologic evaluations included in vitro autoradiography in rat brain and PET scans on anesthetized cats. Results: The chemical and radiochemical purities of 18F-F13714 were greater than 98%. F13714 has a high affinity (0.1 nM) and selectivity for 5-HT1A receptors. In vitro 18F-F13714 binding in rats was consistent with the known 5-HT1A receptors distribution (hippocampus and cortical areas) and was particularly high in the dorsal raphe. In vitro binding of 18F-F13714 was blocked in a dose-dependent fashion by WAY100635, the prototypical 5-HT1A antagonist, and by the endogenous agonist, serotonin (5-HT). Addition of Gpp(NH)p also inhibited in vitro 18F-F13714 binding, consistent with a preferential binding of the compound to G-protein–coupled receptors. Ex vivo tissue measurements in rat revealed an absence of brain radioactive metabolites. In vivo studies showed that the radiotracer entered the cat brain readily and displayed a preferential labeling of 5-HT1A receptors located in cingulate cortex. In vivo labeling was prevented by preinjection of WAY100635. Conclusion: 18F-F13714 is a radiofluorinated agonist that presents suitable characteristics for probing the high-affinity states of the 5-HT1A receptors in vitro and in vivo. Thus, it is a promising tool for investigation of 5-HT1A agonist binding in the living human brain.
The implication of serotonin 1A (5-hydroxytryptamine 1A [5-HT1A]) receptors in behavior and processes underlying emotion has been established in several animal models with species-dependant mechanisms (1). It is also believed to be involved in the pathophysiology of major neuropsychiatric disorders such as schizophrenia, Parkinson disease, and depression (2).
The functional imaging of 5-HT1A receptors by PET may provide further insight into the role of these receptors in pathologic states and treatment assessment. Currently, several selective 5-HT1A antagonist PET ligands are available for mapping and quantifying 5-HT1A receptor binding in the human brain (3–6).
It is known that G-protein–coupled receptors, such as the 5-HT1A receptor, exist in a high- and a low-affinity state when they are coupled or uncoupled to their G proteins, respectively (7,8). Whereas 5-HT1A antagonists bind to the total pool of receptors, 5-HT1A agonists bind preferentially to the high-affinity state of the receptor (9–11). Therefore, differences between the magnitude of binding of antagonist and agonist radiotracers in a given central nervous system area may give an indication about the proportion of G-protein–coupled 5-HT1A receptors, possibly providing us with a more accurate representation of the population of functional receptor (12). Moreover, agonist tracers may be more sensitive to extracellular changes in 5-HT because they should both compete for receptors in the high-affinity state (13).
To date, all PET studies in patients have been performed with 5-HT1A antagonist radiotracers, most commonly 11C-WAY100635 and 4-(2′-methoxyphenyl)-1-[2′-(N-2″-pyridinyl)-p-18F-fluorobenzamido]ethylpiperazine (18F-MPPF). Therefore, it is of great interest to develop a radiolabeled 5-HT1A agonist for in vivo studies in animals and humans.
Although several agonist candidates have been investigated in vitro, their use in vivo has largely remained unsuccessful (14–18). Most failures were attributed to insufficient specificity or affinity of the radiotracers for 5-HT1A receptors. Three teams, including ours, recently reported the preclinical evaluation of 11C-labeled agonist tracers in rats and primates (19,20) and of an 18F-labeled agonist tracer in rats and cats (21). Although some of these radiotracers provided encouraging results, their pharmacologic properties were far from ideal. An initial study reported the successful use of CUMI-101, an 11C-labeled agonist tracer, in rats, nonhuman primates, and humans. Even if modeling parameters of this radiotracer were thoroughly explored for 5-HT1A imaging (22–24), its pharmacologic properties were recently questioned because 11C-CUMI-101 exhibits only partial agonism at 5-HT1A receptors (25,26).
In this context, we chose a highly selective 5-HT1A agonist (F13714) that displays subnanomolar affinity for 5-HT1A receptors (27). In this article, we discuss the radiosynthesis, in vitro autoradiographic, and in vivo PET studies of 18F-F13714 as a prerequisite for future use as a PET ligand in humans.
MATERIALS AND METHODS
Synthesis of 18F-F13714 Nitro Precursor
3-Chloro-4-Nitrophenyl-(4-Fluoro-4-{[((5-Methyl-4-Methyl-Tertbutoxycarbonylamino)-Pyridin-2-Ylmethyl)-Tertbutoxycarbonylamino]-Methyl}-Piperidin-1-yl)-Methanone (2)
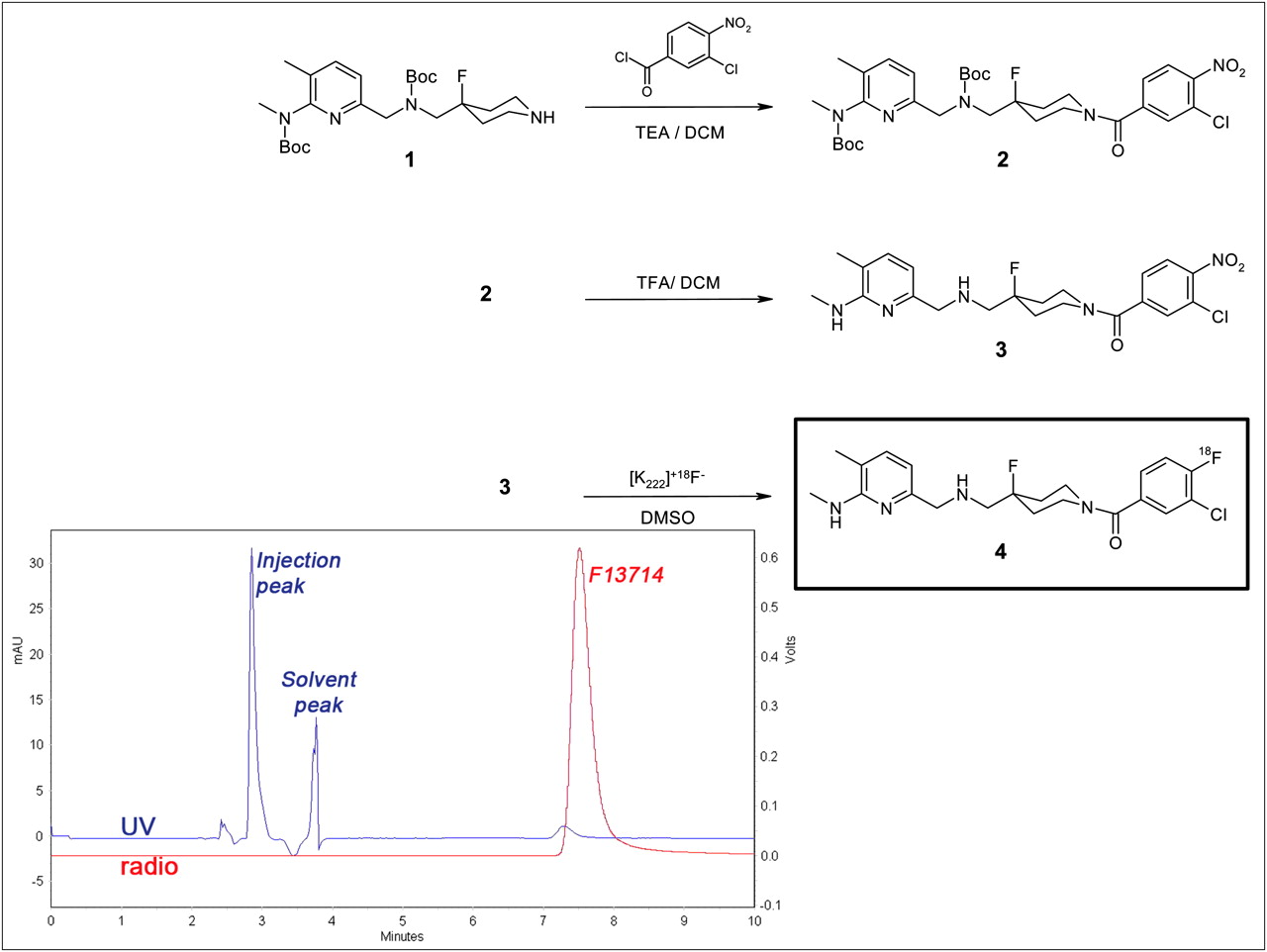
To a stirred solution of 3-chloro-4-nitrobenzoic acid (1.2 g, 5.96 mmol) in dichloromethane (20 mL) and dimethylformamide (0.2 mL), cooled at 0°C, was added dropwise oxalyl chloride (0.5 mL). The mixture was stirred for 2.5 h at room temperature then added dropwise to a solution of 4-fluoro-4-{[(5-methyl-4-methyl-tertbutoxycarbonylamino-pyridin-2-ylmethyl)-tertbutoxycarbonylamino]-methyl}piperidine 1 (2.5 g, 5.36 mmol) in dichloromethane (50 mL), triethylamine (1 mL, 7.15 mmol), and dimethylaminopyridine (catalytic amount) maintained at 0°C (Fig. 1). The temperature was then allowed to rise to room temperature, and stirring was continued for 20 h. The reaction was quenched by addition of water (80 mL) and decanted, and the organic layer was dried on magnesium sulfate and then concentrated under reduced pressure. The residue was purified by flash-column chromatography (dichloromethane-methanol, 95:5) to give 2.45 g (70%) of 2. 1H nuclear magnetic resonance (NMR) (400 MHz, deuterated chloroform [CDCI3], δ): 1.37 (s, 8H), 1.48 (s, 2H), 1.61–1.96 (m, 8H), 2.05 (s, 3H), 3.00 (m, 3H), 3.13–3.74 (m, 7H), 4.13 (s, 1H), 4.44–4.53 (m, 4H), 6.32 (d, J = 7.2 Hz, 1H), 7.14 (d, J = 7.2 Hz, 1H), 7.42 (dd, J = 8.4 Hz and J = 1.2 Hz, 1H), 7.59 (d, J = 1.2 Hz, 1H), 7.91 (d, J = 8.4 Hz, 1H); mass spectrometry (MS) (electrospray ionization [ESI]) m/z = 650 [MH+].

Scheme for synthesis of 2, and 3 and radiosynthesis of 18F-F13714. Quality chromatogram of final 18F-F13714 (ultraviolet [UV] absorbance at 254 nm [UV in blue] and radioactivity [radio in red]) shows radiochemical purity > 98%. DCM = dichloromethane; DMSO = dimethylsulfoxide; TEA = triethylamine.
3-Chloro-4-Nitrophenyl-(4-Fluoro-4-{[((5-Methyl-4-Methyl-Amino)-Pyridin-2-Ylmethyl)-Amino]-Methyl}-Piperidin-1-yl)-Methanone (3)
To a stirred solution of 2 (2.43 g, 3.74 mmol) in dichloromethane (30 mL), cooled at 0°C, was added trifluoroacetic acid (7 mL). The temperature was then allowed to rise to room temperature, and stirring was continued for 24 h. The reaction mixture was diluted with dichloromethane, and then an aqueous solution of sodium hydroxide (1N) was added. The mixture was decanted, the aqueous layer was extracted with dichloromethane, and the combined organic layers were dried on magnesium sulfate and then concentrated under reduced pressure. The residue was purified by flash-column chromatography (dichloromethane-methanol, 95:5) to give 1.4 g (83%) of 3. 1H NMR (400 MHz, CDCl3, δ): 1.58–2.18 (m, 8H), 2.80 (m, 2H), 3.02 (d, J = 4.4 Hz, 3H), 3.19 (t, J = 11.8 Hz, 1H), 3.47 (m, 2H), 3.76 (s, 2H), 4.17 (m, 1H), 4.55 (m, 1H), 6.44 (d, J = 7.2 Hz, 1H), 7.14 (d, J = 7.2 Hz, 1H), 7.42 (dd, J = 8 Hz and J = 1.2 Hz, 1H), 7.59 (d, J = 1.2 Hz, 1H), 7.91 (d, J = 8 Hz, 1H); MS (ESI) m/z = 450.1 [MH+]; analysis calculated for C21H25N5O3ClF: C, 56.06; H, 5.60; N, 15.57; found: C, 56.18; H, 5.40; N, 15.59. High-performance liquid chromatography (HPLC) purity was 98.5% (Xbridge [Waters] C8 5 μm, 1 mL/min, ultraviolet 220 nm, acetonitrile-water-KH2PO4, 250:750:6.8 g, pH 4, with a retention time of 10.5 min).
Radiosynthesis of 18F-F13714 and Quality Control
18F was obtained via an 18O(p,n)18F reaction (IBA Cyclone 18/9 cyclotron). The nitro/fluoro exchange was realized on a standard coincidence synthesizer (TracerLab Mx; GE Healthcare) after the automation sequence was reprogrammed: after initial fluoride preparation (collection, drying, and Kryptofix [Sigma-Aldrich] activation), 4 mg of F13714 nitro precursor (3) were introduced with 3 mL of dimethyl sulfoxide, and the reaction mixture was heated at 150°C for 10 min. After dilution with 15 mL of water, the reaction mixture was passed through an activated C18 cartridge for prepurification, and the crude product was eluted from the cartridge with 1 mL of methanol. Pure 18F-F13714 was obtained after separation on a preparative HPLC column (C18 Symmetry Prep; 7 μm, 7.8 × 300 mm; Waters) eluted with 20 mM H3PO4/tetrahydrofuran/trifluoroacetic acid 88%/12%/0.1% at 3 mL⋅ min−1, with a retention time of 45 min (Fig. 1). For biologic use, the radiotracer was formulated via solid-phase extraction techniques. The product was diluted with 40 mL of sterile water, and the loaded Sep-Pak Light C18 cartridge (Waters) was eluted with 1 mL of ethanol; the final product was diluted with isotonic saline and sterilized by filtration (sterile filter Millex-GS, 0.22 μm; Millipore). Finally, 5% ethanol in saline-injectable solution was obtained.
Quality control consisted of determining radiochemical purity and specific activity, by analytic HPLC assay (ultraviolet set at 254 nm and radioactive detection; C18 Nucleodur 5 μm, 4.6 × 250 mm column [Macherey-Nagel]; elution with 20 mM H3PO4/tetrahydrofuran/MeOH 77.5/9/13.5 at 1 mL⋅min−1, with a retention time of 7 min) of an aliquot of the labeled product by comparing it with a calibration curve generated from solutions of known concentration.
Partition Coefficient Measurement of 18F-F13714
Lipophilicity (log D) was measured to evaluate the lipid solubility of 18F-F13714 by partitioning between n-octanol and 50 mmol/L Tris-HCl (pH 7.4) buffer. Log D was taken as the concentration of 18F-F13714 in n-octanol over the concentration in buffer.
Drugs and Animals
F13714, a selective 5-HT1A receptor agonist, was synthesized by the Pierre Fabre Research Center. 8-OH-DPAT hydrochloride, another selective 5-HT1A receptor agonist, and WAY-100635, a selective 5-HT1A receptors antagonist, were obtained from Sigma-Aldrich. Gpp(NH)p, a nonhydrolysable analog of guanosine 5′-triphosphate, was purchased from Sigma-Aldrich, and urethane was obtained from Acros Organics.
Adult male Sprague-Dawley rats (Charles River Laboratories; weighing 250–300 g) and a European male cat (weighing 3.5 kg) were used. All animals were kept in the animal facility (room temperature, 22°C, with a 12-h day–night cycle). All experiments were realized in accordance with European guidelines for care of laboratory animals (86/609 EEC) and were approved by the ethical animal use committee of the Université de Lyon.
In Vitro Autoradiographic Studies
After a short inhalation of isoflurane, rats were decapitated and brains were carefully removed and immediately frozen in 2-methylbutane cooled with dry ice (−29°C). Coronal sections (30 μm thick) across the hippocampus, raphe nuclei, and cerebellum were cut using a −20°C cryostat, thaw-mounted on glass slides, and allowed to air dry before storage at −80°C until used. The day of 18F-F13714 synthesis, the slides were allowed to reach ambient temperature and were then incubated for 20 min in Tris phosphate-buffered saline buffer (138 mM NaCl, 2.7 mM KCL, pH adjusted to 7.6) containing 18F-F13714 (37 kBq/mL [1 μCi/mL]). For competition experiments, the slides were placed in the same buffer supplemented with WAY100635 (1, 10, and 100 nM), F13714 (1, 10, and 100 nM), Gpp(NH)p (10 μM), or 5-HT (1 nM, 10 nM, 100 nM, and 1 μM). After incubation, the slides were dipped in cold buffer (4°C) for 90 s, in distilled cold water (4°C) for 90 min, and then dried and juxtaposed to a phosphor imaging plate for 60 min (BAS-1800 II; Fujifilm). Region of interest (cingulate cortex, ventral hippocampus, dorsal raphe nucleus, and cerebellum) were drawn manually using Multigauge software (Fujifilm). The results were expressed in arbitrary units or in percentage of control.
Brain Determination of Unchanged Radiotracer
Three series of 4 rats were anesthetized by intraperitoneal injection of urethane (1.7 g/kg) and a catheter was placed into their caudal vein. The rats were killed by decapitation 10, 20, 30, and 40 min after a bolus injection of 18F-F13714 (55.5 MBq [1.5 mCi]). The brains were rapidly removed; the hippocampus was dissected, homogenized with an ultrasonic homogenizer in 100 μL of perchloric acid (0.4 mol/L), and centrifuged at 1,000g for 10 min. The supernatant was neutralized by 30 μL of 4 M potassium acetate and filtered (0.45 μm) before HPLC. The HPLC system consisted of a C-18 reversed-phase column (Nucleodur [Macherey-Nagel], a 7.8 × 300 mm column, and elution with 20 mM H3PO4/tetrahydrofuran/MeOH 77.5/9/13.5) at a flow rate of 1 mL/min. During elution, 2-min fractions were collected and counted for radioactivity with an automated γ-counter (Cobra II; Packard).
In Vivo Cat Brain Studies
Cat PET Scan Acquisition
The PET images were acquired for 2 European male cats anesthetized with 5% isoflurane (Baxter) for 5 min; anesthesia was then lowered to 2.5%. The cat was placed in an acrylic stereotactic apparatus with ear bars, and a catheter was inserted into the forearm branch of the brachiocephalic vein continuously perfused with NaCl 0.9%. The PET scan was acquired on a CTI Exact ECAT HR+ (Siemens) used in 3-dimensional mode. During the first 10 min of acquisition, the transmission scan was obtained with 3 rotating 68Ge and 68Ga sources, followed by a 74 MBq (2 mCi) injection of 18F-F13714. The radioactivity was measured in a series of 31 sequential frames of increasing duration from 30 s to 10 min. The total duration of the scan was 90 min. The images obtained after a duplicate experiment were reconstructed following the frontal plan. Regions of interest (cingulate cortex [500 mm3], hippocampus [450 mm3], and cerebellum [3,200 mm3]) were manually drawn with the software CAPP (CTI-Siemens) according to the stereotactic atlas of the cat brain (12,28,29). The raphe nucleus was not distinguished because of the spatial resolution of the PET camera. The radioactivity of different regions attributed to the fixation of 18F-F13714 was expressed in Bq/cm3. In another experiment with the same animal, WAY-100635, a 5-HT1A antagonist, was administered (1 mg/kg intravenously) 30 min before the injection of 18F-F13714 (74 MBq intravenously). The PET scan acquisition and the image analysis were performed in a similar manner.
Cat MR Image Acquisition
The same cats were anesthetized with 5% isoflurane for 5 min, and then the anesthesia was lowered to 2.5%. The head of the animal was immobilized in a stereotactic acrylic frame with ear bars and orbital and hard palate pieces. MR image acquisitions (1.5-T Magnetom scanner; Siemens AG) consisted of a 3-dimensional anatomic T1-weighted sequence. The anatomic volume covered the whole brain, with 0.7 mm3 voxels.
RESULTS
Synthesis of Nitro Precursor and Radiosynthesis of 18F-F13714
The synthetic route for the preparation of F13714 is shown in Figure 1. Synthesis of 3-chloro-4-nitrophenyl-(4-fluoro-4-{[(5-methylpyrimidin-2-ylmethyl) tertbutoxycarbonylamino]-methyl}-piperidin-1-yl)-methanone (2) was achieved from 3-chloro-4-nitrobenzoic acid and substituted piperidine (1). Removal of the Boc protecting group provided the precursor (3) in high purity. Labeling of 18F-F13714 (4) was obtained from (3) with a radiochemical yield of 10% corrected for decay and 90-min radiosynthesis time using a reprogrammed automated fluorination module. No radioactive by-products were observed, and the HPLC conditions chosen ensured good separation of 18F-F13714 from its nitro precursor, as confirmed by quality control (Fig. 1). Radiochemical purity was greater than 98%, and specific activity ranged between 80 and 150 GBq⋅μmol−1, corrected at EOS.
Partition Coefficient Measurement
The lipophilicity of 18F-F13714, as determined by the octanol-water partition coefficient (log D), was 2.2 (average of 3 experiments)—a value in agreement with the calculated log D (version 7.0 software; ACD/Labs).
In Vitro Distribution Studies
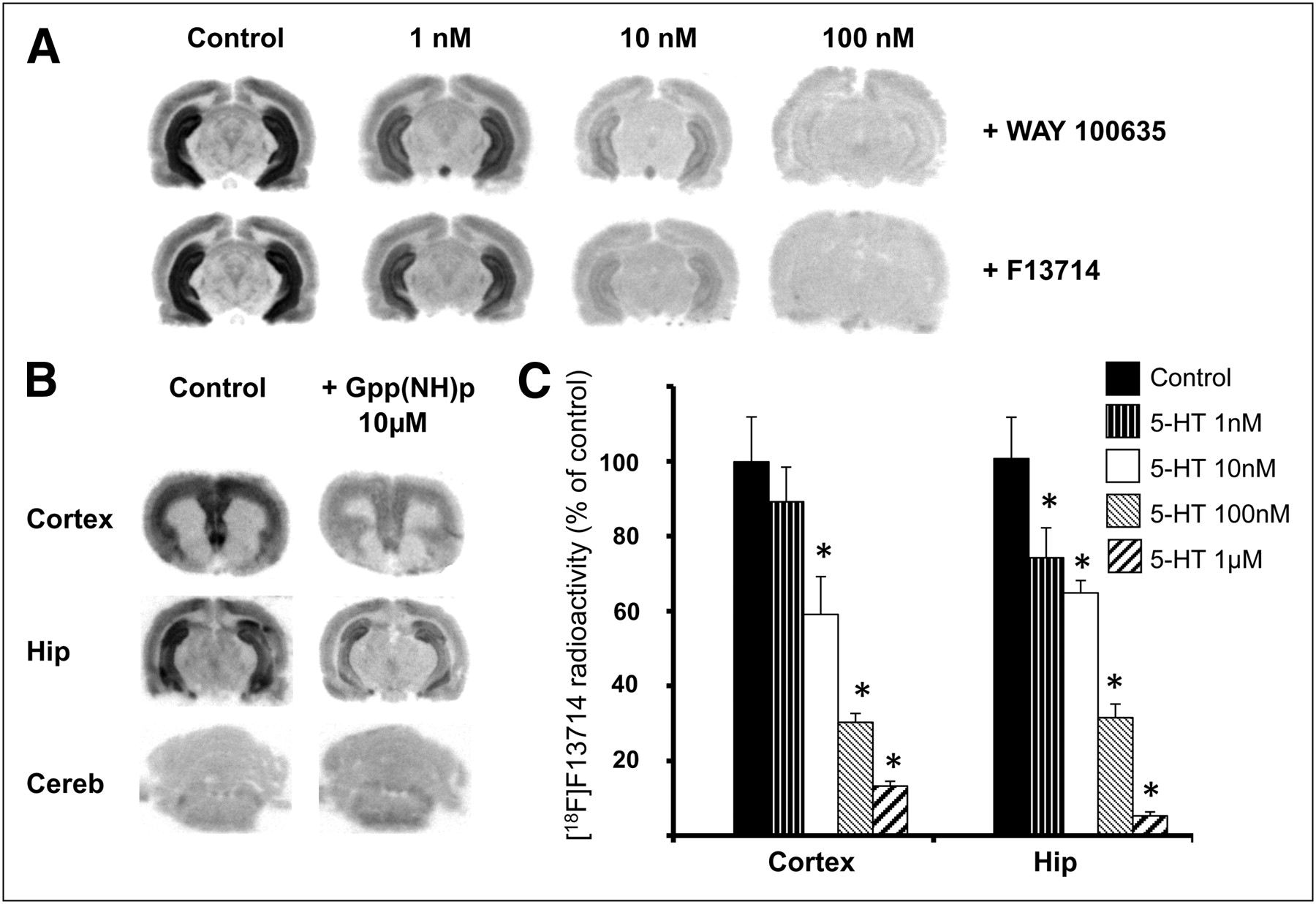
The distribution of 18F-F13714 labeling was evaluated by semiquantitative autoradiography in rat brains (Fig. 2). The labeling distribution pattern obtained in vitro after incubation at a constant concentration of 18F-F13714 fitted with that observed with 18F-MPPF, giving a clear preference for labeling 5-HT1A receptors in specific regions such as the cingulate and entorhinal cortex, hippocampus, raphe nucleus, and, to a lesser extent, brain stem. No binding was observed in the cerebellum, a region with a low expression of 5-HT1A receptors. Specific binding of 18F-F13714 (ratio of the region of interest to cerebellum) was 5.5 in the hippocampus and 3.3 in the dorsal raphe.
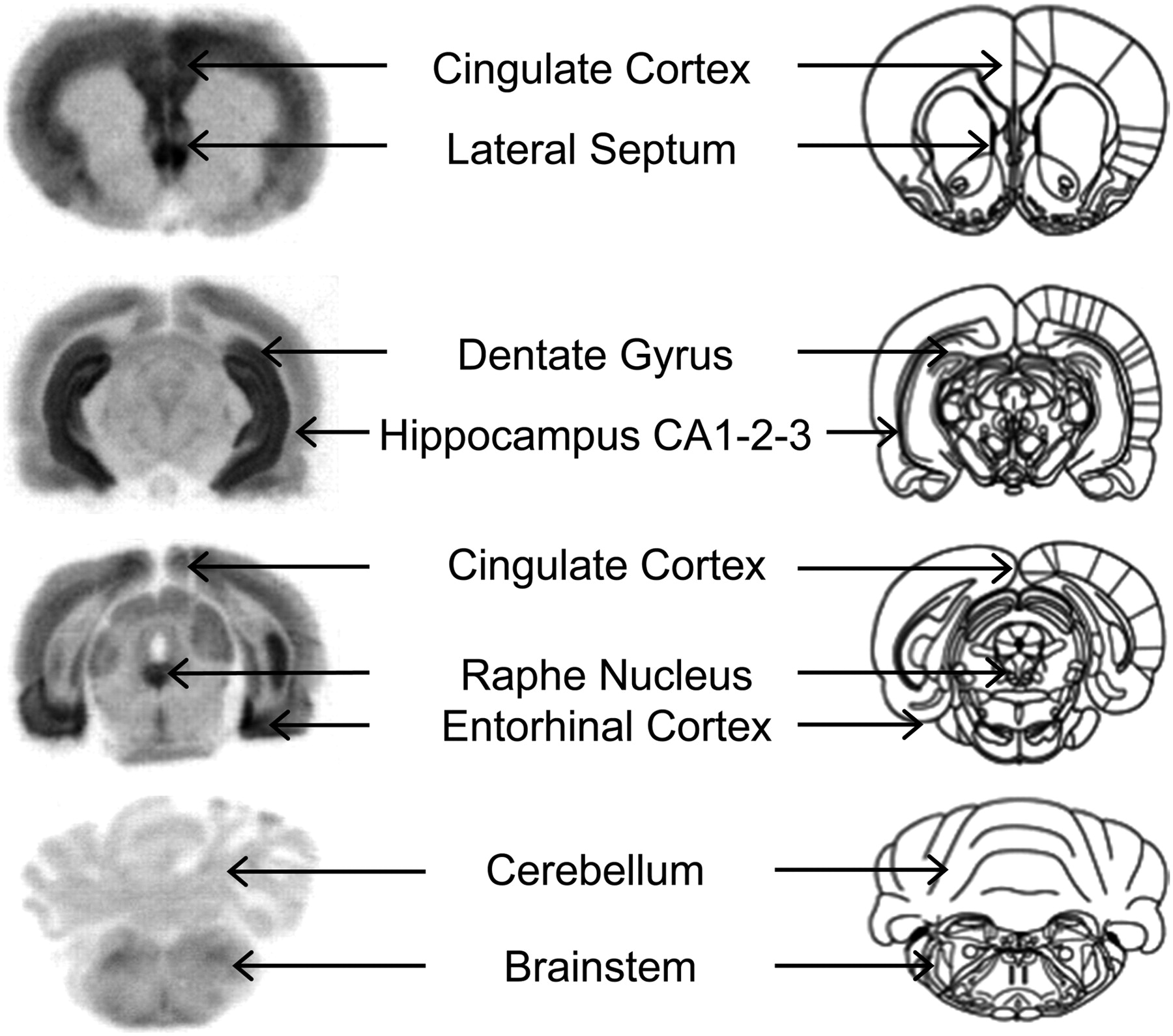
In vitro autoradiograms of brain sections of rat incubated with 18F-F13714. Corresponding anatomic slices show radioactive binding locations corresponding to that of 5-HT1A receptors.
In Vitro Competition Studies
In the hippocampus, 18F-F13714 radioactivity measured by displacement experiments (performed in triplicate) was markedly reduced after the addition of WAY100635, a 5-HT1A receptor antagonist (−20%, −66%, and −83% with 1, 10, and 100 nM of WAY100635, respectively, Fig. 3A). The addition of cold F13714 at the same concentrations reduced binding of 18F-F13714 by 28%, 77%, and 85%, respectively (Fig. 3A), providing further evidence of the high affinity of F13714 for 5-HT1A receptors. In other experiments, the incubation with 10 μM Gpp(NH)p, a nonhydrolysable analog of guanosine 5′-triphosphate that elicits uncoupling of receptors from G proteins, reduced the 18F-F13714 binding by 70% in the cortex and 60% in the hippocampus, with no modification in the cerebellum (Fig. 3B). At increasing concentrations of 5-HT, 18F-F13714 binding decreased by 10% and 20% with 1 nM, 40% and 30% with 10 nM, 70% and 70% with 100 nM, and 90% and 95% with 1 μM, in the cortex and hippocampus, respectively (Fig. 3C).
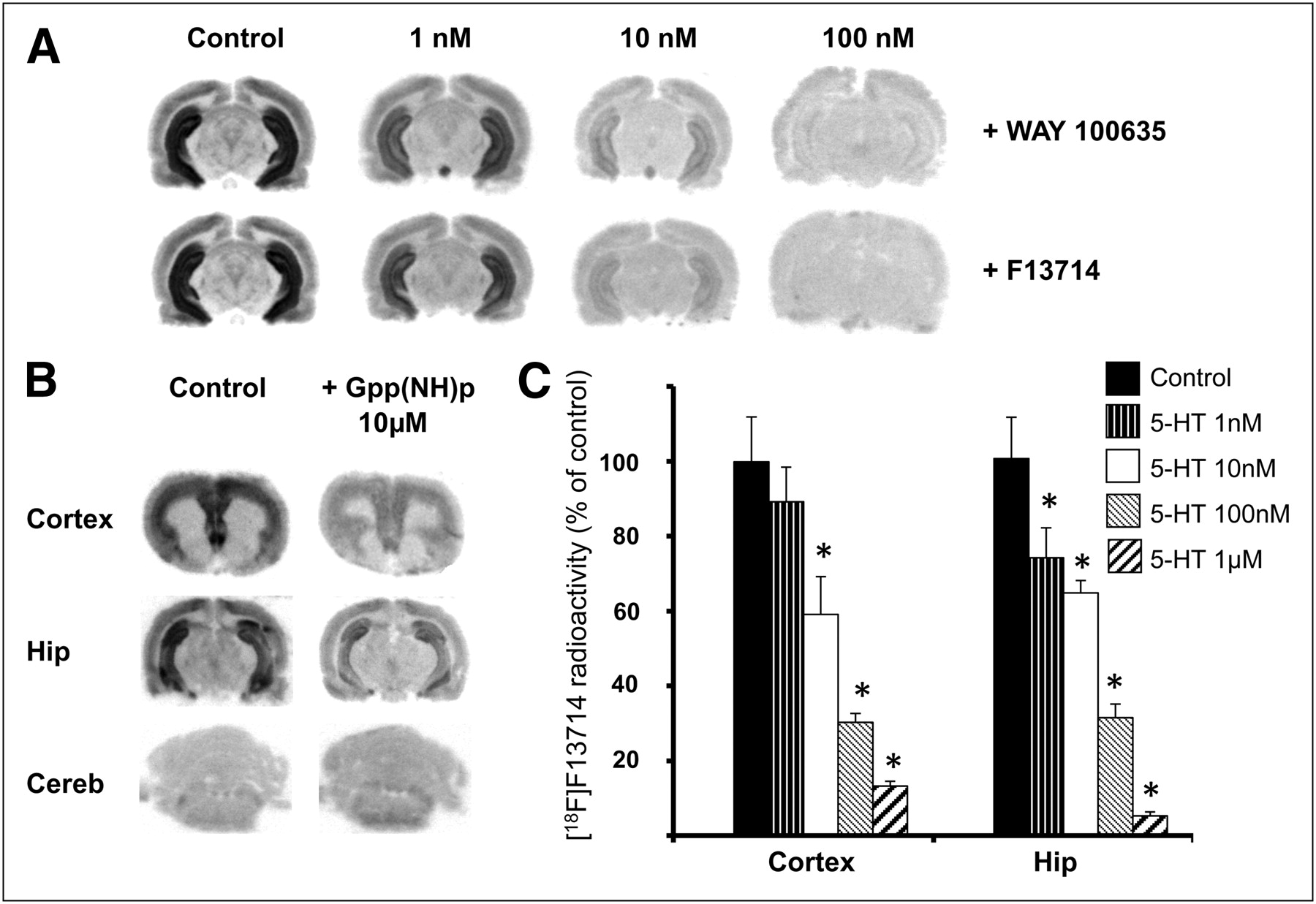
(A) In vitro autoradiography in rats (in hippocampus) and competition between 18F-F13714 and 5-HT1A antagonist WAY-100635, or unlabeled F13714, at different concentrations. (B) In vitro autoradiography in rats and measures of effect of 10 μM of Gpp(NH)p, decoupling receptors from G proteins in cortex, hippocampus (Hip), and cerebellum (Cereb). (C) Histogram obtained by in vitro autoradiography in rats and competition between 18F-F13714 and increasing concentrations of serotonin. Radioactivity measured in cortex or hippocampus is in percentage of control. Mean values (+SEM) are significantly different (*P < 0.05) when comparing with controls using unpaired Student t test.
Unmetabolized Radiotracer in Rat Brain
In rat brain over the time scale considered (10, 20, 30, and 40 min after injection), most of the radioactivity came from unchanged 18F-F13714 (80%, 90%, 94%, and 91%, respectively, for experiments performed in triplicate).
In Vivo 18F-F13714 Binding in Cat Brain
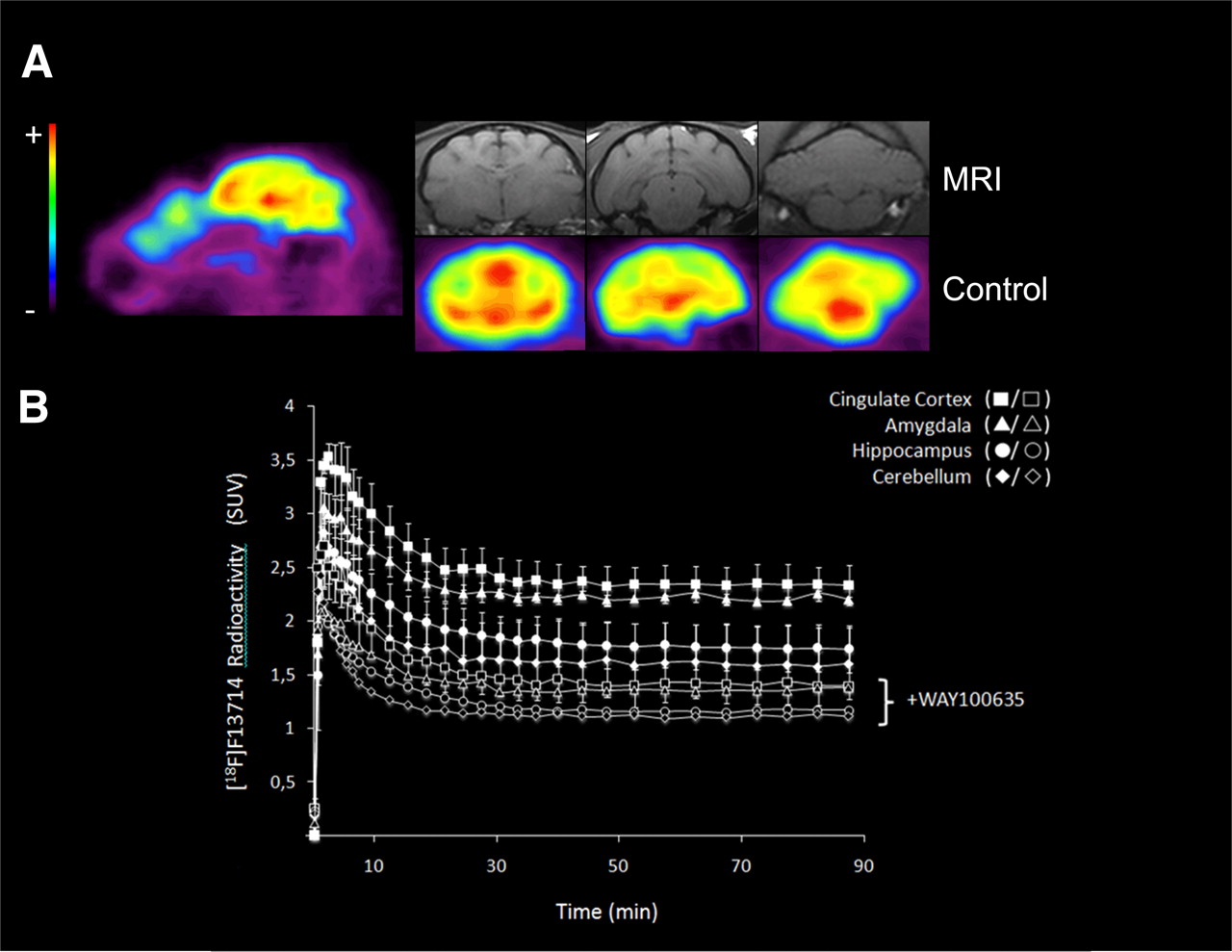
PET images from the cat brain showed 18F-F13714 binding in the 5-HT1A-rich regions (i.e., cingulate cortex, amygdala, and hippocampus), whereas no binding was observed in 5-HT1A-poor regions such as the striatum and cerebellum (Fig. 4A). The time–activity curves showed a high uptake in the hippocampus, cingulate cortex, and cerebellum several minutes after injection of the radiotracer (Fig. 4B). The binding ratio was 1.5 for cingulate cortex to cerebellum, 1.4 for amygdala to cerebellum, and 1.2 for hippocampus to cerebellum (experiments performed in duplicate). It is striking that labeling was higher in the cortex than in the hippocampus, in which the density of 5-HT1A receptors is higher. The injection of WAY100635 before that of the radiotracer significantly decreased 18F-F13714 binding in all areas (experiments performed in duplicate). Thus, in the presence of WAY100635 (1 mg/kg intravenously), the binding ratio was 1.2 for cingulate cortex to cerebellum, 1.2 for amygdala to cerebellum, and 1 for hippocampus to cerebellum.
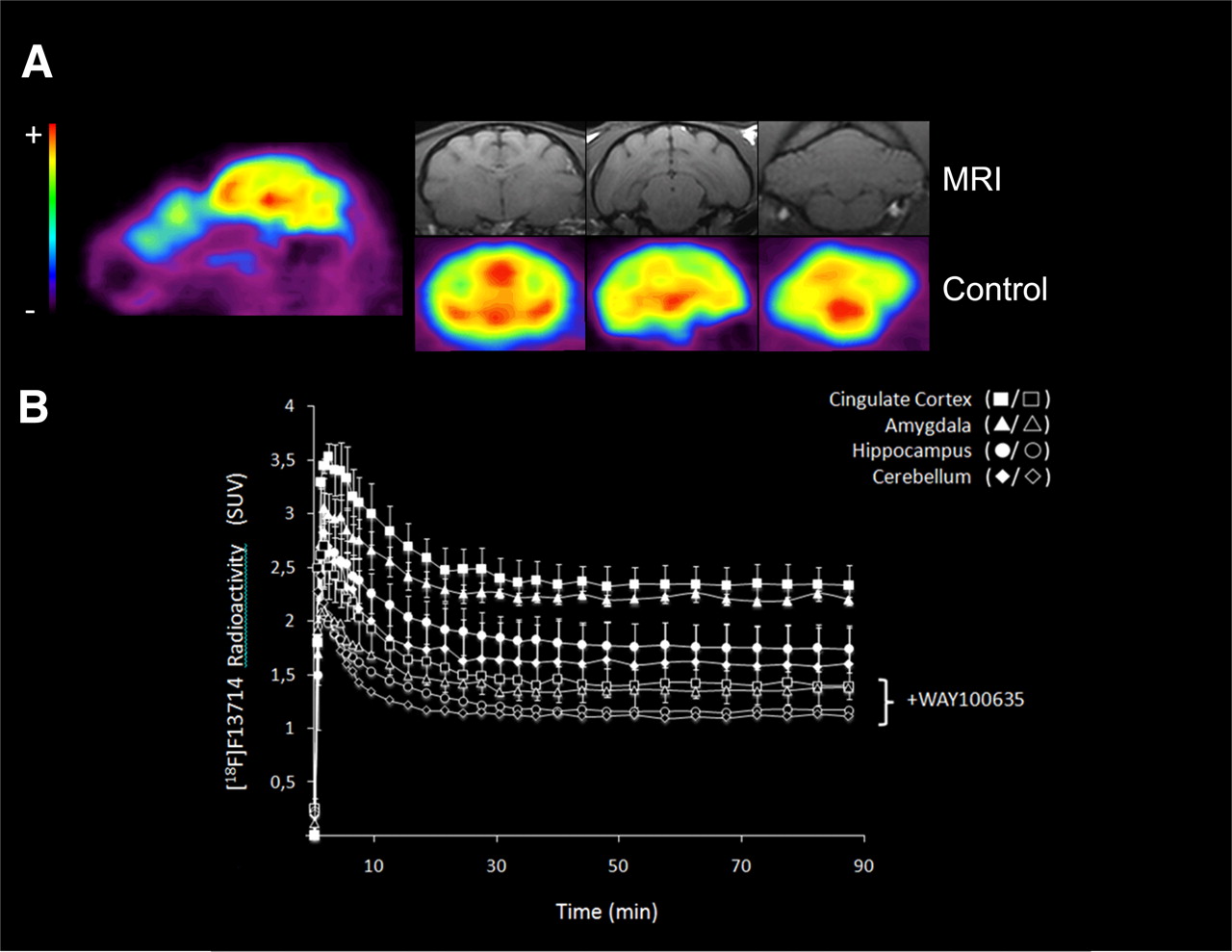
(A) Representative sagittal and coronal PET images summed for 0–90 scans, showing 18F-F13714 distribution in cat brain (and corresponding MR images). Pseudocolor scale from low (blue, −) to high binding level (red, +). (B) Time–activity curves of 18F-F13714 at baseline (solid symbols) and after intravenous WAY100635 blockade (open symbols), 5-HTIA antagonist (1 mg/kg, 30 min before radiotracer injection, duplicated experiments).
DISCUSSION
All currently validated 5-HT1A radiopharmaceuticals for clinical PET are antagonists, and many efforts have already been devoted to developing an agonist radioligand to explore functional 5-HT1A receptors. We recently radiolabeled a pharmacologically well-characterized agonist, F15599, which exhibits a high selectivity for 5-HT1A receptors and a distinctive preferential agonism at postsynaptic 5-HT1A receptors (21). Even though the radiopharmacologic profile of 18F-F15599 allowed in vivo imaging of G-protein–coupled 5-HT1A receptors, the labeling intensity was deemed insufficient for development as a radiopharmaceutical (21). The limited labeling intensity of in vivo images was ascribed to the insufficient affinity of 18F-F15599 for 5-HT1A receptors (apparent inhibition constant [Ki], 2.24 nM). Because 5-HT1A receptor density (Bmax) is about 6 nM in rat hippocampus (including G-protein–coupled and uncoupled receptors), a radioligand affinity lower than 1 nM would be desirable to achieve a higher-contrast image.
Accordingly, we investigated a new 5-HT1A agonist, F13714, that would present several advantages as a PET radiotracer. First, like its chemical analog F15599, F13714 exhibits an outstanding selectivity for 5-HT1A receptors. With the exception of σ-binding sites (inhibitory concentration of 50% [IC50], ∼80 nM), the affinity of F13714 for 40 other receptors, enzymes, and transporters was at least 1,000-fold lower (27,30). Second, its chemical structure carries a fluorine atom amenable to radiofluoration (more convenient for clinical uses than 11C radiolabeling). Third and importantly, F13714 exhibits an exceptionally high affinity for 5-HT1A receptors, with a Ki of 0.1 nM or less—that is, 0.06 nM at rat cortical 5-HT1A receptors, 0.10 nM at rat hippocampal 5-HT1A receptors, and 0.04 nM at recombinant human 5-HT1A receptors expressed in Chinese hamster ovary cells (30–32). Its remarkable agonist efficacy leads some authors to consider agonists from this chemical series as superagonists (33). The high affinity of F13714 is crucial because the specific binding of a radiotracer is a function of its affinity for the site relative to the density (Bmax) of that binding site. Therefore, an affinity lower than 1 nM would be desirable to achieve a good-contrast image for 5-HT1A receptors (34).
From a chemical standpoint, the labeling from the nitro precursor of 18F-F13714 was straightforward and proceeded with excellent radiochemical purity and satisfactory specific activity. The calculated lipophilicity predicted a good blood–brain barrier penetration (35) and was confirmed by the favorable in vivo central nervous system uptake measured in cat PET scans. In tests aimed at evaluating brain metabolism, we found that more than 90% of brain radioactivity was due to unchanged radiotracer (between 10 and 40 min after injection), which allowed us to discard any significant contribution from radiolabeled metabolites to specific binding.
In vitro distribution of 18F-F13714 labeling in rat brain sections was consistent with the reported distribution of 5-HT1A receptors in the hippocampus, dorsal raphe nucleus, and cortical areas (36). Interestingly, in vitro binding in the dorsal raphe nucleus was more pronounced for 18F-F13714 than for 18F-F15599 (34) and similar to 18F-MPPF (37). This higher labeling intensity of (presynaptic) 5-HT1A autoreceptors in the dorsal raphe observed with 18F-F13714 was linked to its ability to discriminate between 5-HT1A receptors coupled to different G-protein subtypes (both subtypes Gi and Go for 18F-F13714, whereas 18F-F15599 preferentially activated Gi in vitro). Indeed, coimmunoprecipitation studies have indicated that 5-HT1A receptors preferentially couple to distinct G-protein subtypes in different brain regions: Gi in the dorsal raphe and other G-protein subtypes in other brain regions (38). Correspondingly, it has previously been shown that F13714 exhibited a distinctive signal transduction signature in vitro (39), leading to a preferential activation of presynaptic versus postsynaptic 5-HT1A receptors. Thus, it is hypothesized that the capacity of agonist radiotracers to label 5-HT1A receptors in different brain regions is associated with preferential interactions at receptors that couple to different G-protein subtypes.
In vitro displacement studies confirmed the specificity of 18F-F13714 for 5-HT1A receptors as binding in the hippocampus was markedly reduced after addition of unlabeled WAY-100635. Clearly, the vast majority of F13714 labeling in the brain represented binding to 5-HT1A receptors. The low level of nonspecific binding observed with 18F-F13714 and the negligible amount of brain radioactive metabolites allowed, therefore, a sensitive delineation of 5-HT1A receptors.
The agonistic behavior of F13714 has to be preserved in its radiolabeled form for it to be of any use as a radiotracer. In theory, an agonist tracer is more sensitive to extracellular changes in the endogenous agonist (5-HT in this case) because they both compete with receptors in the high-affinity state (13). The particular sensitivity of 18F-F13714 we observed for low concentrations of 5-HT in vitro (1–10 nM) goes in this direction. Accordingly, 18F-F13714 binding decreases in the presence of Gpp(NH)p, a nonhydrolysable guanosine 5′-triphosphate analog that switches the receptor into an uncoupled, low-affinity state for agonists (14). This observation demonstrates the agonistic behavior of 18F-F13714 at least in vitro but remains to be confirmed in vivo—that is, by pharmacologic challenges increasing extracellular 5-HT.
Our previous studies in cats using the agonist 18F-F15599 showed a preferential binding of this compound to cortical areas relative to the hippocampus. This finding was striking because the density of 5-HT1A receptors is higher in the hippocampus than in the cortex (36). Indeed, Bmax values of 5-HT1A receptors (determined by autoradiography in rats with 3H-WAY100635) in the hippocampal formation, entorhinal cortex, and dorsal raphe are reported to be between 187 and 243, 194, and 157 fmol/mg of wet tissue weight, respectively (40). An obvious question is whether there is a potential labeling by 18F-F13714 with non–5-HT1A binding sites. However, the high selectivity of both compounds for 5-HT1A receptors seems to rule out interactions with other targets. As mentioned, all the labeling seen with 18F-F13714 was inhibited in vitro by coincubation with WAY-100635 and in vivo by preinjection of WAY-100635. Cat brain PET labeling with 18F-F13714 was particularly marked in the cortex and amygdala, whereas it was modest in the hippocampus and undetectable in the raphe nucleus. Such a distribution of labeling does not correspond to the expression levels of the receptor in these different brain tissues (higher in the hippocampus than in the cortex) and is clearly different from that seen with 18F-MPPF (12). The results with 18F-F13714 are similar to those reported previously with 18F-F15599 (34) but with a much higher contrast image in favor of 18F-F13714. Taken together, the differences observed here in the labeling patterns of comparison of 18F-F13714 and 18F-F15599 provide further evidence that these agonists distinguish 5-HT1A receptor subpopulations in different brain areas. We are confident that the radiopharmacologic profile of 18F-F13714 will be appropriate for clinical studies, taking into account its well-characterized pharmacology (27,30,39). A quantitative approach using kinetic models is now needed to complete this work. In this regard, arterial input measures are currently in progress.
CONCLUSION
This study has shown that 18F-F13714 binds specifically to high-affinity 5-HT1A receptors and has also demonstrated the feasibility of using this radioligand to visualize functional 5-HT1A receptors in rat or cat brains. Our findings establish that F13714 exhibits a high selectivity for 5-HT1A receptors in vitro, where it labels the dorsal raphe, and in the hippocampus and cortex; is sensitive to G-protein uncoupling by guanine nucleotides and to 5-HT in vitro; and binds preferentially to cortical versus hippocampal 5-HT1A receptors in vivo in cats. The innovative profile of 18F-F13714 should therefore open the way to subtle region-specific molecular imaging of 5-HT1A receptors closely implicated in diverse psychiatric or neurologic disorders.
DISCLOSURE STATEMENT
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
No potential conflict of interest relevant to this article was reported.
Footnotes
Published online May 10, 2012.
- © 2012 by the Society of Nuclear Medicine, Inc.
REFERENCES
- Received for publication November 23, 2011.
- Accepted for publication February 28, 2012.





{kind=link}
{kind=link}
{kind=link}
{kind=link}