


Abstract
The development of a PET radioligand for imaging 5-hydroxytryptamine (5-HT) 6 receptors in the brain would, for the first time, enable in vivo imaging of this target along with assessment of its involvement in disease pathophysiology. In addition, such a tool would assist in the development of novel drugs targeting the 5-HT6 receptor. Methods: On the basis of in vitro data, GSK215083 was identified as a promising 5-HT6 radioligand candidate and was radiolabeled with 11C via methylation. The in vivo properties of 11C-GSK215083 were evaluated first in pigs (to investigate brain penetration and specific binding), second in nonhuman primates (to confirm brain penetration, specific binding, selectivity, and kinetics), and third in human subjects (to confirm brain penetration and biodistribution). Results: 11C-GSK215083 readily entered the brain in all 3 species, leading to a heterogeneous distribution (striatum > cortex > cerebellum) consistent with reported 5-HT6 receptor densities and distribution determined by tissue-section autoradiography in preclinical species and humans. In vivo saturation studies using escalating doses of GSK215083 in primates demonstrated saturable, dose-dependent binding to the 5-HT6 receptor in the striatum. Importantly, 11C-GSK215083 also exhibited affinity for the 5-HT2A receptor; however, given the differential localization of these 2 receptors in the central nervous system, the discrete 5-HT6 binding properties of this radioligand were able to be determined. Conclusion: These data demonstrate the utility of 11C-GSK215083 as a promising PET radioligand for probing the 5-HT6 receptor in vivo in both preclinical and clinical species.
The development of selective PET radioligands has facilitated the in vivo imaging of a range of targets in the central nervous system. Of these, the serotonergic system has received much attention with the development of PET radioligands for the 5-hydroxytryptamine (5-HT) 1A (1), 5-HT1B (2), 5-HT2A (3), and 5-HT4 receptors (4), along with the 5-HT transporter (5). However, efforts to produce useful radiotracers for other serotonergic proteins, in particular the 5-HT6 receptor, have had limited success (6).
The 5-HT6 receptor is 1 of 14 distinct mammalian serotonin receptors expressed in the central nervous system, through which serotonin is involved in the regulation of several biologic processes (7). The role of the 5-HT6 receptor in the human brain is not yet clearly understood. However, its localization in the basal ganglia and limbic regions and the high affinity of several atypical antipsychotics suggest that this receptor subtype may participate in the serotonergic control of motor function, mood-dependent behavior, depression, and cognition. Preclinical studies have demonstrated that 5-HT6 receptor antagonism can exert cognitive enhancing effects, in part by inducing increases in the extracellular levels of neurotransmitters in the frontal cortex (8). These properties suggest a potential utility for 5-HT6 receptor antagonists in the treatment of cognitive and behavioral problems characteristic of diseases such as dementia and Alzheimer disease.
The development of high-affinity, selective 5-HT6 receptor antagonists has enabled the study of these receptors in greater detail. Tissue-section receptor autoradiography studies have characterized the binding and distribution of the radioligands 125I-SB-258585 and 3H-Ro-63-0563 in various species (9,10). These studies have consistently revealed heterogeneous binding throughout the brain, with the highest levels found in the striatum, moderate levels in the cerebral cortex, and the lowest levels in the cerebellum. Cross-species comparisons of 5-HT6 receptor distribution have shown similar distribution patterns between rats and humans (11). However, little information is known about 5-HT6 receptor distribution in large preclinical species. Equilibrium saturation studies in rat, pig, and human striata have revealed respective receptor densities of 173 ± 22, 181 ± 25, and 215 ± 41 fmol/mg of protein (12).
Efforts to develop PET radioligands suitable for in vivo 5-HT6 receptor imaging have had limited success, since candidates displaying both good brain penetration and sufficient specific binding signal have been difficult to identify (6). Recently, a new generation of antagonist chemotypes has become available (13). The promising brain penetration properties of these novel quinolines led to a search for derivatives that could be labeled with either 11C or 18F and evaluated as potential radioligands. GSK215083 was identified as a lead candidate for radiolabeling with 11C. GSK215083 had a lipophilicity profile favorable for brain penetration (distribution coefficient [log D], 2.0–2.5) and exhibited a subnanomolar affinity (Ki) for the human recombinant 5-HT6 receptor stably expressed in the HeLa cell line (0.16 nM; Table 1) over other receptor subtypes, with the exception of 5-HT2A, for which GSK215083 demonstrated an affinity approximately only 5-fold lower (Table 1). These findings have led to the evaluation of GSK215083 as the first PET radioligand for the 5-HT6 receptor. This paper reports the radiosynthesis of 11C-labeled GSK215083, its in vivo evaluation as a 5-HT6 PET radioligand in pigs and nonhuman primates (NHPs), and its initial assessment in humans.
In Vitro Binding Profile of GSK215083
MATERIALS AND METHODS
Radiosynthesis of 11C-GSK215083
GSK215083 and the N-desmethyl precursor were prepared according to published procedures (13,14). All other chemicals and solvents were of analytic grade and were used without further purification.
In vivo PET studies were conducted at 3 PET facilities: the Aarhus PET Centre, in Denmark (pig imaging); the Columbia University PET Facility, in the United States (NHP imaging); and the PET Centre of the Centre for Addiction and Mental Health (CAMH), in Canada (human imaging). For all radiosyntheses, the methylation agent 11C-methyliodide was used. 11C-methyliodide was prepared by catalytic reduction of 11C-carbon dioxide to 11C-methane followed by gas-phase iodination with iodine with a PET-Trace system (GE Healthcare) at the Aarhus PET Centre and by reduction of 11C-carbon dioxide to 11C-methanol with lithium aluminum hydride followed by iodination with hydriodic acid at the Columbia University PET Facility and the PET Centre of CAMH.
11C-GSK215083 was prepared by reacting the free base of the corresponding desmethyl precursor (1 mg) with 11C-methyltriflate in the presence of 2,2,6,6,-tetramethylpiperidine (10 μL) at 80°C in a 2:1 mixture of methanol:acetonitrile (300 μL). After 5 min of heating, the reaction mixture was diluted with a solution of the mobile phase (500 μL) and injected onto a semipreparative column (SphereClone ODS(2) C-18, 250 × 10 mm [Phenomenex]; mobile phase, acetonitrile:70 mM NaH2PO4 (60:40); flow rate, 9 mL/min) for high-performance liquid chromatography (HPLC) purification. Fractions eluting between 10 and 12 min were combined, evaporated to dryness, and reformulated using saline. The reformulated product was analyzed for radiochemical purity and specific activity by radio-HPLC (SphereClone C-18, 250 × 4.6 mm; acetonitrile:70 mM NaH2PO4 (70:30); flow, 2 mL/min; retention time, 4.8 min). The lipophilicity (measured log D) of 11C-GSK215083 was determined using the method of Wilson et al. (15).
Metabolite Profiling Studies and Dosimetry Estimation in the Rat
Both the metabolite profiling and the dosimetry studies were performed at Huntingdon Life Sciences.
For metabolite studies, Lister hooded male rats (254–292 g; n = 10) were administered 3H-GSK215083 (0.4 μg/kg). 3H-GSK215083 was radiochemically pure (99.4%), with a specific activity of 3.07 TBq/mmol. Brain samples were collected at 5 time points between 5 and 120 min after administration (n = 2 animals per time point). Samples were pooled and extracted twice with acetonitrile with sonication. Supernatants were combined and aliquots taken for scintillation counting. Combined extracts were reduced and diluted with water:acetonitrile:trifluoroacetic acid (800:200:1; 1 mL). Samples were spiked with nonradiolabeled GSK215083 before injection onto the HPLC column (Hypersil BDA C-18, 250 × 4.6 mm [Thermo Scientific]; mobile phase, A = 0.1% trifluoroacetic acid in water, B = 0.1% trifluoroacetic acid in acetonitrile; flow rate, 1 mL/minute) for online radioactivity and ultraviolet detection at 254 nm (room temperature). The retention time of nonradiolabeled GSK215083 was 15 min.
Rat dosimetry was performed to determine the expected radiation dose to human male subjects after administration of radiolabeled GSK215083. Briefly, a single dose of 3H-GSK215083 was administered to male Lister hooded rats. Animals were sacrificed at 5 time points between 5 and 120 min after administration of the radioligand, and 29 different tissues were assessed for biodistribution of 3H-GSK215083.
In Vivo Pig PET Studies
Pig studies were performed in accordance with the Danish Animal Experimentation Act at the Aarhus PET Centre. Yorkshire/Danish Landrace pigs (40 kg; n = 2) were kept fasting for 24 h before PET examination, with free access to water.
Homologous competition was investigated in pigs by means of 4 sequential PET scans, using high-specific-activity intravenous administrations of 11C-GSK215083 with escalating doses of unlabeled GSK215083. After a baseline 11C-GSK215083 scan, 3 further scans were performed on the same animal in the course of a day, with intravenous administration (1-min bolus) of escalating doses of unlabeled GSK215083 (0.005, 0.05, and 0.5 mg/kg) 5 min before injection of the radioligand.
After initial intramuscular induction of anesthesia with midazolam and ketamine, the animals were intubated onto a respirator with medical-grade air. Anesthesia was maintained with isoflurane (1%–2%) for the duration of the study. Catheters were surgically placed into the left femoral vein and artery of the animal for administration of radiopharmaceuticals and pharmaceutical agents and for blood sampling and blood pressure recording. Throughout the study, vital signs were monitored and maintained within the normal physiologic range.
Pigs were positioned in the PET scanner (ECAT EXACT HR tomograph; Siemens) and immobilized using a custom-made head-holding device. After a transmission scan for the purpose of subsequent attenuation correction, dynamic PET data were acquired over the brain in 3-dimensional mode with the animal supine. Emission data were acquired for 90 min after intravenous injection of 11C-GSK215083. Each scan consisted of a sequence of 26 frames (8 × 15 s, 4 × 30 s, 2 × 1 min, 2 × 2 min, 4 × 5 min, 6 × 10 min). Measured attenuation and scatter corrections were applied to the emission data. Data were reconstructed using the reprojection algorithm (16) with an axial and transaxial Hanning filter that had a 0.5 cutoff frequency. The resulting images had a spatial resolution of approximately 5–7 mm in full width at half maximum (17).
During each acquisition, forty 2 mL blood samples were collected from the femoral artery for the determination of the whole-blood and plasma input functions. The radioactivity concentrations were measured using a γ-counter (Cobra II Auto-Gamma; PerkinElmer).
The fraction of radioactivity in pig plasma corresponding to authentic radioligand was determined by radio-HPLC. Arterial blood samples (seven 2-mL samples obtained between 2 and 60 min) were obtained after administration of the radioligand. After centrifugation (10,000 rpm, 1 min), isolated plasma (∼0.5 mL) was treated with acetonitrile (∼0.5 mL) and centrifuged again (10,000 rpm, 5 min). After the protein had been removed from the plasma samples, they were spiked with unlabeled GSK215083 (∼10 μg) and analyzed by radio-HPLC (SphereClone ODS(2) C-18, 250 × 10 mm; mobile phase, acetonitrile:70 mM NaH2PO4 (70:30); flow rate, 2 mL/min).
Plasma measurements of radioactivity concentration were fitted with a model that consisted of a linear interpolation before the peak and a sum of 3 exponentials after the peak. This curve was corrected by the parent fraction model, which consisted of an exponential plus a constant fitted to the parent fraction measurements,
A pig brain atlas was registered to the 0- to 90-min summed PET image via an affine transformation. The registered brain atlas was applied to the PET dynamic data to generate time–activity curves for several brain regions, including the striatum, frontal cortex, and cerebellum.
In Vivo NHP PET Studies
All data were acquired under protocols approved by the Columbia University Medical Center Institutional Animal Care and Use Committee. In total, 6 NHPs (Papio anubis) were scanned.
For each study day, 2 intravenous administrations of 11C-GSK215083 were administered to an NHP. The first scan was a baseline high-specific-activity scan, and the second scan followed administration of a dose of unlabeled GSK215083 (0.0002–0.5 mg/kg), 30 min before injection of the radioligand. Twelve scans were obtained (6 at baseline and 6 after administration of unlabeled GSK215083).
The fasting animals were immobilized with ketamine (10 mg/kg, intramuscularly) and anesthetized with isoflurane (2%) via an endotracheal tube. A venous cannula was inserted for hydration and tracer delivery. A cannula was inserted in the femoral artery for blood sampling. Vital signs were monitored, and body temperature was maintained at 37°C with heated water blankets.
PET data were acquired on an HR+ scanner (Siemens). Each PET scan was 120 min long. After correction of attenuation via a transmission scan, dynamic data were acquired in 3-dimensional mode over the brain. Emission data were acquired for 91 min after intravenous administration of 11C-GSK215083. Each 91-min scan consisted of 19 frames (4 × 15 s, 2 × 1 min, 4 × 2 min, 2 × 5 min, 7 × 10 min). After corrections for decay, attenuation, randoms, scatter, and dead time, emission data were reconstructed by filtered backprojection using a Shepp filter (0.5 cycles per ray).
Arterial plasma was sampled with an automated fraction collection system for the first 4 min (1 every 20 s during the first 2 min and 1 every 30 s during minutes 3 and 4), followed by manual draws at longer intervals for the remainder of the scan. Six additional samples were collected between 2 and 70 min for metabolite analysis via HPLC.
Parent fractions were fitted to a sum of 2 exponentials, with the slowest exponential constrained so that the terminal parent plasma and cerebellum washout were equal,
PET data were coregistered to T1-weighted MRI scans acquired previously for each NHP. Regions of interest were manually drawn on the MRI scans to include the cerebellum, cortex, and striatum. Coregistration was performed by maximization of mutual information. Regions of interest were applied to coregistered dynamic PET data, and time–activity curves were derived.
In Vivo Human PET Studies
The human study was performed after regulatory approval at the PET Centre of CAMH (study number 257/2006). Healthy male subjects (n = 4; 40 ± 2 y old; 78 ± 15 kg [mean ± SD]) were used in this study. Each subject provided written informed consent, and the study was approved by the local research ethics board and Health Canada. Safety data, in terms of medical history, physical examination, electrocardiogram, and vital signs, were collected at screening and at the follow-up visit (within 7 d of the scan). For the PET study, each subject was cannulated in the antecubital vein for tracer administration and in the contralateral radial artery for blood sampling.
Each subject received a single baseline PET scan after an intravenous administration of 11C-GSK215083 to determine in vivo biodistribution of this radioligand in the brain. The subjects were placed supine in the tomograph, with the head immobilized in a head-holding device. For each subject, a low-dose CT transmission scan was obtained to acquire data for attenuation correction. Dynamic data were then acquired in list mode using a Biograph HiRez XVI PET camera system (Siemens Molecular Imaging) (19). Recording of emission data began on bolus intravenous injection of 11C-GSK215083 (352 ± 12 MBq, 3.38 ± 1.69 μg). For all scans, the list-mode data were converted into a sequence of 26 frames (8 × 15 s, 3 × 1 min, 5 × 2 min, 5 × 5 min, 5 × 10 min), for a total of 90 min. For each 3-dimensional sinogram, the data were normalized and attenuation and scatter were corrected before Fourier rebinning was applied to convert the 3-dimensional sinograms into 2-dimensional sinograms. The 2-dimensional sinograms were reconstructed into image space using a 2-dimensional filtered backprojection algorithm, with a ramp filter at the Nyquist cutoff frequency. After reconstruction, a gaussian filter of 5 mm in full width at half maximum was applied and the images calibrated to Bq/cm3. Images were combined to create a single dynamic dataset on which regions of interest were described and time–activity curves derived.
A T1-weighted MRI scan was obtained for each subject (1.5-T Signa Excite HD; GE Healthcare) for coregistration to the human PET data and identification of discrete regional volumes (resolution, 1 × 1 × 1 mm). Dynamic PET data were corrected for motion via frame-to-frame image registration and aligned with the structural T1 MRI scan using SPM5 (Wellcome Trust Centre for Neuroimaging) with a mutual-information cost function.
Kinetic Analysis
One- and two-tissue compartmental models were fitted to the regional tissue time–activity curve obtained from pig and NHP studies using in-house software written in Matlab (The MathWorks). These models included a fixed blood volume contribution (0.05), and data were fitted by the method of weighted least-squares. Model order was assessed using the Akaike information criteria, allowing for the identification of the optimal compartmental structure. The outcome measures of interest were the delivery of the radioligand into brain tissue (K1; mL/min1/cm3) and the total tissue volume of distribution (VT; mL/cm3).
The dose-dependent change in the primate data was analyzed further by fitting a single-site competition model to the VT estimates,
RESULTS
Radiosynthesis of 11C-GSK215083
Injection-ready 11C-GSK215083 was prepared about 40 min from the end of bombardment with a radiochemical yield of 35% ± 5%. The specific activity was 414 ± 143 GBq/μmol (n = 15) for pigs and NHPs and 47.4 ± 20.3 GBq/μmol (n = 4; mean ± SD) for humans at the end of synthesis, uncorrected for decay, following the reaction conditions depicted in Figure 1 (radiochemical purity > 99%). The identity of the radiolabeled material was confirmed by coinjection with a sample of authentic GSK215083, which, under the same elution conditions, showed an identical retention time. Results from 3 independent log D measurements gave a value of 2.25 ± 0.11, confirming that 11C-GSK215083 could offer an appropriate signal-to-noise ratio, in vivo.

Synthetic route for radiolabeling 11C-GSK215083.
Metabolite Profiling Studies and Dosimetry Estimation in the Rat
Metabolite profiling in the rat using intravenous doses of 3H-GSK215083 yielded data consistent with the hypothesis that metabolites of GSK215083 are not detected in the central nervous system but can be detected in the periphery.
Data acquired from dosimetry and toxicology studies allow for administration of up to 370 MBq and a mass dose of less than 10 μg of 11C-GSK21503 to human subjects. This mass dose of 11C-GSK215083 (<10 μg) represents a microdose and is covered by toxicology studies (GSK, unpublished data, 2005). Radiodosimetry data indicate that a dose of 370 MBq of 11C-GSK215083 produces a committed effective dose exposure of 8.22 × 10−3 mSv/MBq.
In Vivo Pig PET Studies
A representative baseline image and corresponding time–activity curves for 11C-GSK215083 uptake into the pig brain are given in Figures 2A and 3A, respectively. 11C-GSK215083 readily entered the brain, with the highest uptake observed in the striatum (caudate and putamen), moderate uptake in the cortical regions, and the lowest uptake in the cerebellum. Peak radioactive concentrations were observed approximately 10–20 min after administration of 11C-GSK215083, followed by a slow washout from the striatal region and increased washout rates from the cortical and cerebellar regions.

Baseline PET images showing biodistribution of 11C-GSK215083 in pig (A), NHP (B), and human (C) brain (scale bar = %ID/L).

Representative baseline time–activity curves of 11C-GSK215083 in pig (A), NHP (B), and human (C) brain.
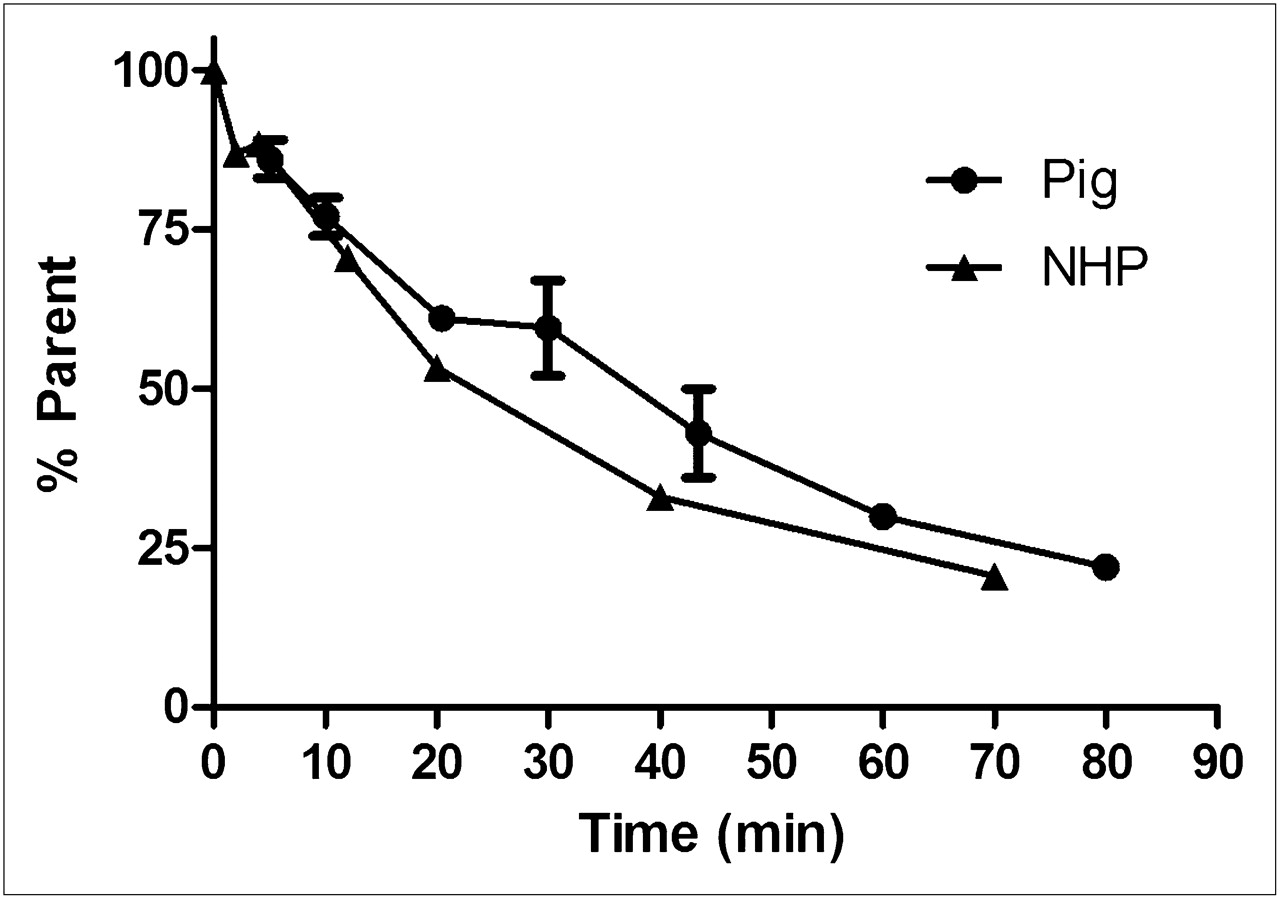
Radio-HPLC analysis revealed 11C-GSK215083 to be rapidly metabolized in arterial plasma, with the parent compound representing about 50% of the total radioactivity 35 min after injection (Fig. 4). In addition to the parent compound, only polar radiometabolites were observed, and these would not be expected to penetrate the blood–brain barrier, consistent with observations made in rat brain.

Blood metabolite profile of 11C-GSK215083 in pig and NHP (mean ± SEM).
The regional time–activity curves were well described by both the reversible one-tissue compartmental model and the reversible two-tissue compartmental model. Overall, Akaike model selection criteria did not consistently indicate a preference for one or two tissue compartments. Therefore, the one-tissue compartmental model was chosen on the basis of prudence and identifiability criteria. VT values were highest in the putamen (∼25 mL/cm3) and lowest in the cerebellum (∼15 mL/cm3).
The rank order of regional VT for 11C-GSK215083 from the baseline scan was striatum > cortex > cerebellum (Table 2), consistent with reported 5-HT6 receptor densities and distribution determined by tissue-section autoradiography (10,11). In vivo saturability studies using 11C-GSK215083 plus increasing doses of unlabeled GSK215083 yielded a dose-dependent decrease in specific binding of 11C-GSK215083 in both the striata and the cortex, with a small decrease in binding also observed in the cerebellum (Table 2; Figs. 5A and 6A). Application of Equation 3 to the VT data provided regional estimates of ED50 of 87.3 μg in the striatum and 148 μg in the frontal cortex. The cerebellum region demonstrated only a small change in VT after administration of pharmacologic doses of unlabeled GSK215083 (ED50, 2118 μg), consistent with the notion of minimal specific binding in this region due to low levels of 5-HT6 and 5-HT2A receptor expression (Fig. 6A).
VT Values (mL/cm3) Generated for Individual Brain Regions from In Vivo Saturation Studies Conducted on Pigs and NHPs

Representative time–activity curves of 11C-GSK215083 before and after administration of blocking dose of unlabeled GSK215083 in pig (0.5 mg/kg) (A) and NHP (0.05 mg/kg) (B), for putamen (5-HT6-rich) and cerebellum (reasonably 5-HT6-devoid) regions of interest.

Plot of in vivo VT vs. log total mass dose of unlabeled GSK215083 (in vivo saturation plots) in pig (A) and NHP (B). Corrected Akaike criteria indicate that, in this primate species, cerebellum is best described by a constant and, in both species, all other regions are best described by a nonlinear saturation model.
In Vivo NHP PET Studies
A representative baseline image and time–activity curves for 11C-GSK215083 uptake into the NHP brain are given in Figures 2B and 3B, respectively. 11C-GSK215083 readily entered the brain, reaching peak levels approximately 10–15 min after administration. Consistent with our findings in pigs, the rank order of regional uptake of 11C-GSK215083 was found to be striatum > cortex > cerebellum, with the slowest washout of 11C-GSK215083 observed from the striatal regions.
The in vivo metabolism of 11C-GSK215083 in the NHP revealed 11C-GSK215083 to be rapidly metabolized in arterial plasma, with the parent compound representing about 50% of the total radioactivity 20 min after injection (Fig. 4).
The regional time–activity curves were well described by both the reversible one-tissue compartmental model and the reversible two-tissue compartmental model. Overall, Akaike-model selection criteria did not consistently indicate a preference for one or two tissue compartments. Therefore, the one-tissue compartmental model was again chosen on the basis of prudence and identifiability criteria. VT values were highest in the putamen (∼17 mL/cm3) and lowest in the cerebellum (∼8 mL/cm3).
Homologous competition with increasing doses of unlabeled GSK215083 yielded a dose-dependent decrease in the VT of 11C-GSK215083 consistent with that observed in the pig (Table 2; Figs. 5B and 6B) in both the striata and the frontal cortex. Application of Equation 3 to the VT data provided regional estimates of ED50 of 10.16 μg in the striata and 55.10 μg in the frontal cortex. There was an approximately 5.4-fold lower value for ED50 observed for the striatal region than for the frontal cortical region, as equates well with the approximately 5-fold higher affinity for 5-HT6 receptors (located mainly in the striatal regions) versus 5-HT2A receptors (located mainly in the cortical regions) measured in vitro for GSK215083 (Table 3). The cerebellum region did not demonstrate any change in VT after administration of pharmacologic doses of unlabeled GSK215083, consistent with the assumption of minimal specific binding in this region due to low levels of 5-HT6 receptor expression (Fig. 6B).
Comparison of In Vitro Human Binding Affinities of GSK215083 with In Vivo Primate ED50 Values
In Vivo Human PET Studies
In humans, no adverse events were reported during the study. A representative baseline image and time–activity curves for 11C-GSK215083 uptake into the human brain are given in Figures 2C and 3C, respectively. 11C-GSK215083 readily entered the brain in all 4 subjects, reaching peak levels approximately 10–20 min after administration. Consistent with our findings in pigs and NHPs, the rank order of regional uptake of 11C-GSK215083 was found to be striatum > cortex > cerebellum, and the slowest washout of 11C-GSK215083 was observed from the striatal regions.
DISCUSSION
This paper describes the radiosynthesis of 11C-GSK215083 and its characterization as a novel PET radioligand for the quantification of central 5-HT6 receptors in vivo in pigs and NHPs, along with an initial assessment in humans.
Screening of compound libraries from a 5-HT6 receptor antagonist program at GlaxoSmithKline identified GSK215083 as a potential 5-HT6 receptor imaging probe that possessed favorable imaging properties. GSK215083 has a lipophilicity range of 1–3 (log D, 2.25 ± 0.11), which confers likely blood–brain barrier penetration. Additionally, GSK215083 possesses a suitable, selective subnanomolar affinity for the 5-HT6 receptor over other protein targets (Table 1), with the exception of the 5-HT2A receptor, for which it also exhibits subnanomolar affinity. This is an important consideration, since the 5-HT6 and 5-HT2A receptors are known to colocalize in several brain regions such as the cortex (10,20), and this characteristic could represent a confounding factor for the use of 11C-GSK215083 in some brain regions. Using data generated from in vitro studies, an estimation of the in vivo nondisplaceable binding potential (BPND) has been derived for both the 5HT-6 receptor and the 5-HT2A receptor, where BPND = (Bmax/Kd) × fND (Bmax is maximum number of binding sites, Kd is affinity, and fND is free fraction in the nondisplaceable compartment) (21). In vitro estimates of the density of 5-HT6 receptors in human striatum (215 fmol/mg of protein (12)), the affinity of GSK215083 for the human 5-HT6 receptor (0.16 nM; Table 1), and the measured free fraction in tissue (fND, 0.0394 (21)) suggested that GSK215083 would possess a suitable signal in vivo (BPND, 5.3). In comparison, the BPND for the 5-HT2A receptors in the striatum is estimated to be 0.64, on the basis of the affinity of GSK215083 for 5-HT2A (0.79 nM; Table 1) and its density in the human striatum (128.5 fmol/mg of protein (20)), suggesting that any contribution of 5-HT2A to the 11C-GSK215083 striatal binding signal would be low. In view of these favorable data, and a successful radiolabeling feasibility assessment, the development of GSK215083 was progressed.
GSK215083 was successfully radiolabeled with 11C with good reproducibility, good radiochemical yields, and high specific activities.
In vivo biodistribution of 11C-GSK215083 in pigs, NHPs, and humans all demonstrated the following rank order of regional brain uptake: striatum > cortex > cerebellum, consistent with reported 5-HT6 receptor densities and distribution determined by tissue-section autoradiography (10,11). In all 3 species, uptake of 11C-GSK215083 was lower in the cerebellum than in any of the other regions investigated. This observation was consistent with in vitro studies reporting low concentrations of 5-HT6 receptors in the cerebellum (10,11).
After administration of increasing blocking doses of unlabeled GSK215083 to pigs and NHPs, a dose-dependent decrease in 11C-GSK215083 VT was observed in the striatal and cortical regions. In contrast, there was no substantial reduction in VT observed in the cerebellum of either pigs or NHPs after administration of unlabeled GSK215083. These data are again consistent with the known 5-HT6 receptor distribution and support the use of the cerebellum as a reference region for this radioligand.
No mass dose effects of GSK215083 were observed when administered at tracer doses to pigs or NHPs (Fig. 6). Using the estimated ED50 for GSK215083 from the NHP striatum (10.16 μg [0.51 μg/kg]) and translating to an average 70-kg human yields a mass dose limit of 3.97 μg such that target occupancy is less than 10% (tracer conditions).
In addition to its affinity for 5-HT6 receptors, GSK215083 is also known to possess a high affinity for 5-HT2A receptors (∼5.4-fold lower than that for 5-HT6; Table 3). Human 5-HT2A receptors are known to exist in high densities in the cortex, moderate densities in the striatum, and low densities in the cerebellum (20,22). The colocalization of 5-HT6 and 5-HT2A receptors in particular areas of the brain, such as cortical regions, implies that binding of 11C-GSK215083 needs careful examination for accurate interpretation of the data.
Saturation studies in the NHP yielded a dose-dependent decrease in specific binding in the striatal and cortical regions of the brain (Fig. 6B). Interestingly, the ED50 for the striatal regions was approximately 5.4-fold higher than that observed in the frontal cortex, as corresponds well with both the known receptor distribution profile of the 5-HT6 and 5-HT2A receptors (i.e., the striatal portion having a high density of 5-HT6 and low density of 5-HT2A receptors and the cortical regions the converse) and the known difference in affinity of GSK215083 for these 2 target proteins (5-fold difference; Table 3).
Given the differences in receptor abundance and binding affinities, we believe that in the NHP, the PET signal exhibited by 11C-GSK215083 is mainly 5-HT6 in the striatal region and mainly 5-HT2A in the cortical region. Therefore, we predict that in the human brain, the binding distribution profile of 11C-GSK215083 for 5-HT6 and 5-HT2A would reflect that observed in the NHP, where binding of the radioligand in the striatum reflects 5-HT6 receptors and binding of the radioligand in the cortex reflects 5-HT2A receptors. For modeling purposes, given the low concentration of both 5-HT6 and 5-HT2A receptors in the cerebellum (10,20) and the relatively unchanged VT values after competition in pigs and NHPs, the use of this region as a reference for modeling PET data generated using 11C-GSK215083 could be considered.
Work is in progress to determine the most appropriate kinetic model for analysis of in vivo PET data using 11C-GSK215083 in humans. Additionally, further studies are under way to characterize this 5-HT6 receptor PET radioligand in humans using known, selective 5-HT2A and 5-HT6 pharmacologic agents. These studies will ultimately determine the utility of 11C-GSK215083 as a PET ligand to assess the 5-HT6 receptor in humans.
CONCLUSION
This paper reports the radiolabeling and pharmacologic investigation of 11C-GSK215083 as a novel 5-HT6 receptor PET radioligand. In vivo biodistribution of 11C-GSK215083 in pigs, NHPs, and humans demonstrated the following rank order of regional brain uptake: striatum > cortex > cerebellum, consistent with reported 5-HT6 receptor densities and localization in preclinical species and humans. Binding of 11C-GSK215083 in pigs and NHPs displayed reversible kinetics. In addition, 11C-GSK215083 has an expected mass dose limit of 3.97 μg in an average 70-kg human, such that target occupancy would be less than 10% (tracer conditions). Further studies are under way to assess the most appropriate kinetic model for analyzing PET data acquired using this radioligand in humans. The data reported in this publication provide evidence that 11C-GSK215083 may be a useful PET radioligand for probing the 5-HT6 receptor in vivo in humans.
DISCLOSURE STATEMENT
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank the staff of the Aarhus PET Centre, the Columbia University PET Facility, and the PET Centre of CAMH (in particular Alan Wilson and Romina Mizrahi) for technical assistance with the pig, NHP, and human studies, respectively. Additionally, we thank Antony Gee, Graham Searle, and Maria Davy for their continued support of these studies over the years. The GlaxoSmithKline-sponsored clinical study was investigator-led and performed at the CAMH on behalf of GlaxoSmithKline. No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Jan. 5, 2012.
- © 2012 by the Society of Nuclear Medicine, Inc.
REFERENCES
- Received for publication May 18, 2011.
- Accepted for publication September 15, 2011.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- An in vivo pig model for testing novel PET radioligands targeting cerebral protein aggregates
- International Union of Basic and Clinical Pharmacology. CX. Classification of Receptors for 5-hydroxytryptamine; Pharmacology and Function
- Age-Related Change in 5-HT6 Receptor Availability in Healthy Male Volunteers Measured with 11C-GSK215083 PET
- A High-Resolution In Vivo Atlas of the Human Brain's Serotonin System
- Human Kinetic Modeling of the 5HT6 PET Radioligand 11C-GSK215083 and Its Utility for Determining Occupancy at Both 5HT6 and 5HT2A Receptors by SB742457 as a Potential Therapeutic Mechanism of Action in Alzheimer Disease