


Abstract
The overexpression and overactivation of hepatocyte growth factor receptor (Met) in various cancers has been linked to increased proliferation, progression to metastatic disease, and drug resistance. Developing a PET agent to assess Met expression would aid in the diagnosis and monitoring of responses to Met-targeted therapies. In these studies, onartuzumab, the experimental therapeutic 1-armed monoclonal antibody, was radiolabeled with 76Br or 89Zr and evaluated as an imaging agent in Met-expressing cell lines and mouse xenografts. Methods: 89Zr-desferrioxamine (df)-onartuzumab was synthesized using a df-conjugate; 76Br-onartuzumab was labeled directly. Met-binding studies were performed using the human tumor–derived cell lines MKN-45, SNU-16, and U87-MG, which have relatively high, moderate, and low levels of Met, respectively. Biodistribution and small-animal PET studies were performed in MKN-45 and U87-MG xenografts. Results: 76Br-onartuzumab and 89Zr-df-onartuzumab exhibited specific, high-affinity Met binding (in the nanomolar range) that was concordant with established Met expression levels. In MKN-45 (gastric carcinoma) xenografts, both tracers cleared slowly from nontarget tissues, with the highest uptake in tumor, blood, kidneys, and lungs. 76Br-onartuzumab MKN-45 tumor uptake remained relatively constant from 18 h (5 percentage injected dose per gram of tissue [%ID/g]) to 48 h (3 %ID/g) and exhibited tumor-to-muscle ratios ranging from 4:1 to 6:1. In contrast, 89Zr-df-onartuzumab MKN-45 tumor uptake continued to accumulate from 18 h (10 %ID/g) to 120 h (23 %ID/g), attaining tumor-to-muscle ratios ranging from 20:1 to 27:1. MKN-45 tumors were easily visualized in imaging studies with both tracers at 18 h, but after 48 h 89Zr-df-onartuzumab image quality improved, with at least 2-fold-greater tumor uptake than nontarget tissues. MKN-45 tumor uptake for both tracers correlated significantly with tumor mass and Met expression and was not affected by the presence of plasma shed Met. Conclusion: 89Zr-df-onartuzumab and 76Br-onartuzumab specifically targeted Met in vitro and in vivo; 89Zr-df-onartuzumab achieved higher tumor uptake and tumor-to-muscle ratios than 76Br-onartuzumab at later times, suggesting that 89Zr-df-onartuzumab would be better suited to image Met for diagnostic and prognostic purposes.
The hepatocyte growth factor receptor tyrosine kinase (Met) is overexpressed or aberrantly activated in a variety of cancers, including gastric, colon, bladder, breast, liver, lungs, pancreatic, esophageal, prostate, and cervical (1). Met gene amplification, mutation, or overexpression in cancer results in increased proliferation, inhibition of apoptosis, tumor angiogenesis, and progression to metastatic disease (1,2). Met amplification or overexpression also has been associated with poor prognosis and drug resistance (3). Together, these attributes suggest that the ability to determine changes in Met expression in real time with a Met-targeted imaging agent could prove invaluable for diagnosis, patient selection, monitoring of disease progression, and identification of instances when a Met-targeted therapeutic might be beneficial for the patient.
Onartuzumab (MetMAb; Genentech (4)), a Met-selective humanized 1-armed monoclonal antibody (∼99,000 kDa; derived from IgG1, monoclonal antibody 5D5) possessing nanomolar affinity for Met, has displayed antiproliferative, antiangiogenic, and proapoptotic properties in mouse tumor models, resulting in inhibition of tumor growth (4,5). Currently, onartuzumab has been studied in phase I–III clinical trials in which it was well tolerated and in which non–small cell lung cancer patients with Met-positive tumors showed improved survival (6). The therapeutic effect of onartuzumab results from competitive antagonism of hepatocyte growth factor binding to the Met extracellular domain, thereby preventing pathway activation (4). Although high affinity and target specificity of onartuzumab are desirable attributes required for a PET agent, the slow in vivo clearance of this large protein from the circulation and nontarget tissues would require the use of longer-lived PET radionuclides, thus ensuring enough time for clearance of nontarget radioactivity but leaving sufficient radioactivity in target regions for detection with PET (7,8). In a previous study by Perk et al. (9), the Met-specific whole monoclonal antibody DN30 (∼150 kDa) labeled with the longer-lived radionuclide 89Zr (t1/2 = 78.4 h) provided semiquantitative images of human Met–expressing tumors in mouse xenografts, proving the utility of longer-lived radionuclides in compensating for the slow pharmacokinetics of large-molecular-weight proteins. In addition, the radiolabeled DN30 tumor uptake corresponded to Met expression levels and represented bivalent binding to Met. Although this bivalent binding was found to result in receptor dimerization and phosphorylation, the ensuing biologic responses were only partial; hence, DN30 has been termed a partial agonist. In contrast, onartuzumab is a pure antagonist capable of only monovalent binding to the receptor, representing quantitatively a 1:1 molar binding ratio (4,10).
For these studies, we have radiolabeled onartuzumab with the longer-lived PET radionuclides 76Br (t1/2 = 16.2 h) or 89Zr and evaluated its imaging potential in vitro and in vivo. 76Br was initially selected because it afforded a direct labeling method with little modification of the antibody whereas 89Zr requires a chelate for conjugation to the antibody. The cell lines MKN-45 (human gastric carcinoma), SNU-16 (human gastric carcinoma), and U87-MG (human glioblastoma)—which have high, moderate, and low Met concentrations, respectively—were used for binding studies to determine the biologic activity (percentage immunoreactivity) and the affinity (dissociation constant, Kd) of 76Br-onartuzumab and 89Zr-desferrioxamine (df)-onartuzumab and in mouse xenograft models to determine the in vivo biodistribution and imaging characteristics of 76Br-onartuzumab and 89Zr-df-onartuzumab (11,12). Additionally, plasma and tumor samples were collected to evaluate human shed Met ectodomain and tumor Met abundance (Met receptor concentration or density), respectively, using immunoassays (11,13). Met sheds its ectodomain (shed Met), a process in which transmembrane portions of the receptor are proteolytically cleaved and released into the biologic circulation. The concentration of shed Met in plasma and urine was found to correlate with tumor burden in mouse xenografts (11). Because shed Met may interfere with binding of radiolabeled onartuzumab, we investigated the relationship between shed Met, tumor Met abundance, and tumor mass. Collectively, these studies demonstrate the potential utility of PET-labeled onartuzumab.
MATERIALS AND METHODS
Cell Lines and Reagents
Cell lines were grown at 37°C and 5% CO2 using RPMI 1640 for MKN-45; RPMI 1640, 10 mM N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid), 1 mM sodium pyruvate, glucose (4,500 mg/L), and sodium bicarbonate (1,500 mg/L) for SNU-16; or Dulbecco modified Eagle medium, 0.1 mM nonessential amino acids, and 1 mM sodium pyruvate for U87-MG. All media were supplemented with 10% fetal bovine serum, 2 mM l-glutamine, and penicillin–streptomycin–amphotericin B.
Onartuzumab that was conjugated to df through an isothiocyanate linker, p-isothiocyanatobenzyl-df-onartuzumab, was provided by Jeffrey Tinianow (Genentech), who used a conjugation procedure previously described (14,15).
Radiosynthesis of 76Br-Onartuzumab and 89Zr-df-Onartuzumab
The radiosynthesis of 76Br-onartuzumab was accomplished by direct bromination of available tyrosine residues. Briefly, 76Br was produced from a 75As (3He, 2n)–76Br reaction and isolated from the solid target by oxidation (16). The radiobrominations were performed with onartuzumab (300 μg) in pH 6.4 phosphate buffer and aqueous ammonium 76Br-bromide (74–185 MBq) in the presence of chloramine-T for 5 min. The product was purified by size-exclusion high-performance liquid chromatography (quadruple time-of-flight mass spectrometry [Waters] coupled with an ultra performance liquid chromatography Acquity system [Waters]). Typical radiolabeling yield was 45% ± 4.4% (n = 10), with a specific activity (at the end of synthesis) of 0.118 ± 0.067 MBq/μg (n = 10).
The radiosynthesis of 89Zr-df-onartuzumab was accomplished using a modified method of Perk et al. (14). Briefly, purified 89Zr (IV) in 1 M oxalic acid (74–148 MBq) was neutralized with Na2CO3 (2 M) and N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (0.5 M, pH 7.0) and reacted with df-onartuzumab (14.7 mg/mL, 0.25 M C2H3NaO2, pH 5.5) in ascorbic or gentisic acid (5 mg/mL) or 1% bovine serum albumin for 1 h. Typical labeling yields determined by size-exclusion high-performance liquid chromatography with ultraviolet monitoring were greater than 90%, with specific activities (at the end of synthesis) ranging from 0.037 to 0.185 MBq/μg.
In Vitro Studies
Saturation binding studies were done to determine the Kd and concentration of Met (Bmax) using plated MKN-45 cells (2 × 105cells per well) and SNU-16 or U87-MG cells in tubes (2–10 × 105cells per tube) to which increasing concentrations of radiolabeled onartuzumab (0.2–10 nM) were added; nonspecific binding was determined by adding unlabeled onartuzumab (10−6.3 M) to another set of duplicates. After incubation for 2 h (4°C), the cell-bound radiolabeled onartuzumab was separated from free onartuzumab. Plated cells were washed (phosphate-buffered saline), treated with trypsin, and collected in vials. Cells in tubes were centrifuged and washed twice (phosphate-buffered saline), and the cell-bound radioactivity for these samples was determined by γ-counting (2480 Wizard3; Perkin Elmer). The Kd and Bmax were determined from at least 6–8 concentrations of radiolabeled onartuzumab and analyzed using nonlinear regression curve fits (1-site binding hyperbola) (Prism, version 3.02 for Windows [Microsoft]; GraphPad Software).
One of 2 methods was used to determine the immunoreactive fractions or biologic specific activity of the radiolabeled onartuzumab as previously described. Briefly, we used a modified method described by Lindmo et al. (17,18) in which the immunoreactivity was calculated by extrapolation to infinite antigen excess conditions. Alternatively, we used a modification of the specific activity determination by self-displacement technique described by Morris (19), which is derived from a radiolabeled onartuzumab saturation curve and competition curve with unlabeled onartuzumab.
Mouse Tumor Models
Athymic female nude mice (Ncr-nu/nu; National Cancer Institute) were injected subcutaneously in the right thigh with MKN-45 or U87-MG cells (5–8 × 106) in phosphate-buffered saline:30% Matrigel (BD Biosciences). All animal studies were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals (20) (National Institutes of Health) using protocols approved by the Institutional Animal Care and Use Committee.
Biodistribution Studies
Nude or tumor (50–1,000 mg)–bearing mice were injected while awake via the tail (intravenously) with either 76Br-onartuzumab (0.185–0.74 MBq, 2–5 μg), 89Zr-df-onartuzumab (0.37–1.85 MBq, 3–46 μg), or 89Zr-oxalate (neutralized) (0.185–0.74 MBq) and euthanized (CO2 inhalation) at selected times. Blood (cardiac puncture) and tissues were excised from each animal and weighed, and the radioactive concentration was determined (2480 Wizard3). The radioactivity in the blood and each tissue was expressed as percentage injected dose per gram of tissue (%ID/g) normalized to a 20-g mouse: 100 × ([cpmtissue]/[cpminjected dose] × [tissue weight (g)]) × ([body weight]/[20]). Immediately after counting, tumor samples were fast-frozen and stored at −70°C for determination of Met content. An additional blood sample was collected in ethylenediaminetetraacetic acid–coated tubes, and after being centrifuged, the separated plasma samples were fast-frozen and stored at −70°C for shed Met analysis.
For the blocking studies with unlabeled onartuzumab, MKN-45 tumor (50–200 mg)–bearing mice were divided into 2 groups, with one group receiving 89Zr-df-onartuzumab (∼0.37 MBq, ∼3 μg) and the other group receiving a coinjection of 89Zr-df-onartuzumab plus 1 mg of unlabeled onartuzumab. The mice were euthanized after 3 d, and the processing of the blood and tissues was performed as described above. Statistical analysis of the differences between the 2 groups was done using the Student t test.
Small-Animal PET Studies
Tumor-bearing mice were anesthetized using isoflurane/O2 (1.5%–3% v/v) and imaged at various times after the intravenous injection of 76Br-onartuzumab (1.85–5.55 MBq, 30–50 μg) or 89Zr-df-onartuzumab (3.7–7.4 MBq, 64–128 μg). Whole-body static images requiring 4–5 bed positions were obtained (field of view, 2.0 cm; total imaging time, 20–25 min) using the Advanced Technology Laboratory Animal Scanner (21). The images were reconstructed using a 2-dimensional ordered-subset expectation maximum from which regions of interest were drawn manually to determine the tissue uptake (kBq/cm3). The %ID/g normalized to a 20-g mouse was then determined using the formula (100 × [tissue uptake]/[injected dose]) × ([body weight]/[20]).
Determination of Plasma Shed Met Ectodomain and Total Tumor Met
Met and plasma Met ectodomain (shed Met) levels were measured using a 2-site electrochemiluminescent immunoassay in which antibodies tagged with a ruthenium chelate (Sulfo-Tag; Meso Scale Discovery) were detected with a SECTOR Imager 2400 plate reader (Meso Scale Discovery) as described previously (11). A purified recombinant Met ectodomain–IgG-Fc fusion protein was used as a reference standard for quantitation of Met mass per mass total extracted cell protein. Tumor tissue extracts were prepared by physically disrupting tissue samples before clearing by centrifugation and analysis of Met content as described above. Correlations of shed Met, total tumor Met, and tumor radioactive onartuzumab content to tumor mass or total tumor mass were performed using the Spearman rank correlation coefficient (Prism)
RESULTS
In Vitro Cell-Binding Studies
Binding studies were performed with 76Br-onartuzumab and 89Zr-df-onartuzumab to determine the Kd, concentration of Met (Bmax, receptors per cell), and the percentage immunoreactive fraction using MKN-45 cells (12). Both 76Br-onartuzumab and 89Zr-df-onartuzumab had high specific binding for Met, ranging from 80% to 96% and from 80% to 98%, respectively. The Kd for 76Br-onartuzumab binding was 5.14 ± 0.81 nM (n = 3), which was comparable to the Kd for 89Zr-df-onartuzumab binding (6.12 ± 1.18 nM; n = 3). The estimated numbers of receptors per cell (calculated from Bmax) were 1.07 ± 0.28 × 106 (n = 3) and 0.936 ± 0.08 × 106 (n = 3) for 76Br-onartuzumab and 89Zr-df-onartuzumab, respectively. The percentage immunoreactive fractions were also comparable, 85.4% ± 6.9% (n = 3) and 81.9% ± 0.7% (n = 3) for 76Br-onartuzumab and 89Zr-df-onartuzumab, respectively. In similar saturation studies with SNU-16 and U87-MG, higher nonspecific binding was observed, ranging from 20% to 30% and from 55% to 85%, respectively, which is consistent with moderate and lower levels of Met expression (11,12). The Bmax was at least 6-fold higher for SNU-16 (0.089 ± 0.013 × 106 [n = 3]) than for U87-MG (0.014 ± 0.003 × 106 [n = 3]). These data compare favorably with the expected levels of Met expression for these human cancer cell lines and further indicate that these tracers would be appropriate for imaging tumors with high to moderate Met expression (>100,000 receptors per cell); however, at lower Met expression levels (<14,000 receptors per cell), nonspecific interactions could impair the detection of specific Met binding.
In Vivo 76Br-Onartuzumab, 89Zr-df-Onartuzumab, and 89Zr-Oxalate
Biodistribution Studies
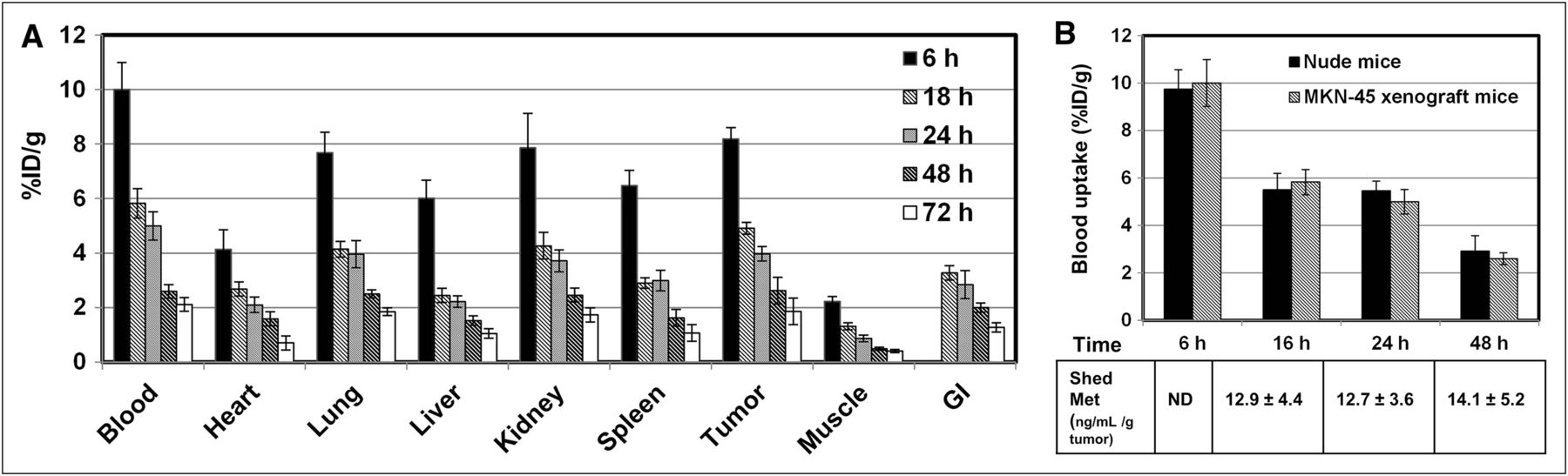
The biodistribution of 76Br-onartuzumab was determined in MKN-45 xenografts from 6 to 72 h (Fig. 1A). The highest uptake was observed in tumor, blood, kidney, and lung, and uptake in tumor was similar to uptake in blood and these nontarget organs at all time points. In similar biodistribution studies using U87-MG xenografts, uptake in nontarget tissues was comparable to that in MKN-45 xenografts, but tumor uptake was approximately 2-fold lower, consistent with the lower Met content of this line. Examining the ratio of %ID/g in the tumor and muscle (a nontarget tissue) to further assess the feasibility of 76Br-onartuzumab as an imaging agent revealed that the highest tumor-to-muscle (TM) ratios, 5:1 to 6:1, occurred after 24 h for MKN-45 xenografts (Table 1). For the U87 xenografts, the TM ratio ranged from 3:1 to 4:1 at 24 and 48 h, respectively, or approximately 1.5- to 2-fold lower, consistent with lower Met expression.
Comparison of TM Ratios of 89Zr-df-Onartuzumab and 76Br-Onartuzumab in MKN-45 Xenografts

(A) Biodistribution of 76Br-onartuzumab in MKN-45 xenografts from 6 to 72 h. Each time point represents mean %ID/g ± SD of 76Br-onartuzumab (n = 3 for 72-h group; n = 4 for all other time points). (B) Comparison of 76Br-onartuzumab blood uptake (%ID/g) in nude and MKN-45 xenograft mice. Each time point represents mean %ID/g ± SD of 76Br-onartuzumab (nude mice, n = 5; MKN-45 xenografts, n = 4). The table shows corresponding plasma shed Met ectodomain concentrations (ng/mL/g tumor) determined from same MKN-45 xenograft mouse blood samples; each value represents mean concentration ± SD (n = 4). GI = gastrointestinal tract; ND = not determined.
Because uptake of 76Br-onartuzumab in blood and tumors was similar over time, studies were undertaken to determine whether the levels of shed Met ectodomain in the blood of tumor-bearing mice differed from that of control mice, potentially influencing the blood clearance. Both of these Met-expressing tumors have been shown to shed Met ectodomain into the systemic circulation (11,12). For these studies, the biodistribution of 76Br-onartuzumab was determined in non–tumor-bearing nude mice after 6, 16, 24, and 48 h and compared with the uptake (%ID/g) shown in Figure 1A for MKN-45 xenografts. At all time points, the %ID/g in blood (Fig. 1B), heart, kidneys, lungs, or spleen was not significantly different (P < 0.05); significant differences were observed in liver uptake at 6 h (28% decrease) and in muscle at 6 h (24% decrease), 16 h (47% decrease), and 48 h (53% increase) in the nude mice versus the xenograft mice. The differences in muscle uptake may be attributable to poor counting statistics because this tissue has relatively low radioactivity. The significant difference in the liver uptake most likely reflects age and body weight differences between the 2 groups. Although the uptake was normalized to a 20-g mouse, this normalization did not take into account age-related changes in metabolism. In this case, the xenograft mice at 6 h were older (∼12 wk) and weighed more (almost 30 g) than the nude mice (age, ∼5 wk; weight, 19–24 g); hence, the younger nude mice would be expected to have faster liver metabolism and clearance than the older xenograft mice, resulting in the observed decreased liver uptake. Further, the concentration of shed Met ectodomain (Fig.1B; ng/mL per gram of tumor mass) in plasma samples obtained from these MKN-45 xenografts remained relatively unchanged at 16, 24, and 48 h, whereas the 76Br-onartuzumab blood uptake steadily decreased. The 76Br-onartuzumab blood concentration (at the time of injection) was approximately 12 nM, which was approximately a 100-fold excess, compared with shed Met blood concentrations of approximately 0.126 nM for a 1-g tumor (representative of the largest tumor and highest shed Met blood concentration). Overall, these results indicate that the presence of shed Met in tumor-bearing mice does not alter the pharmacokinetics and clearance of 76Br-onartuzumab.
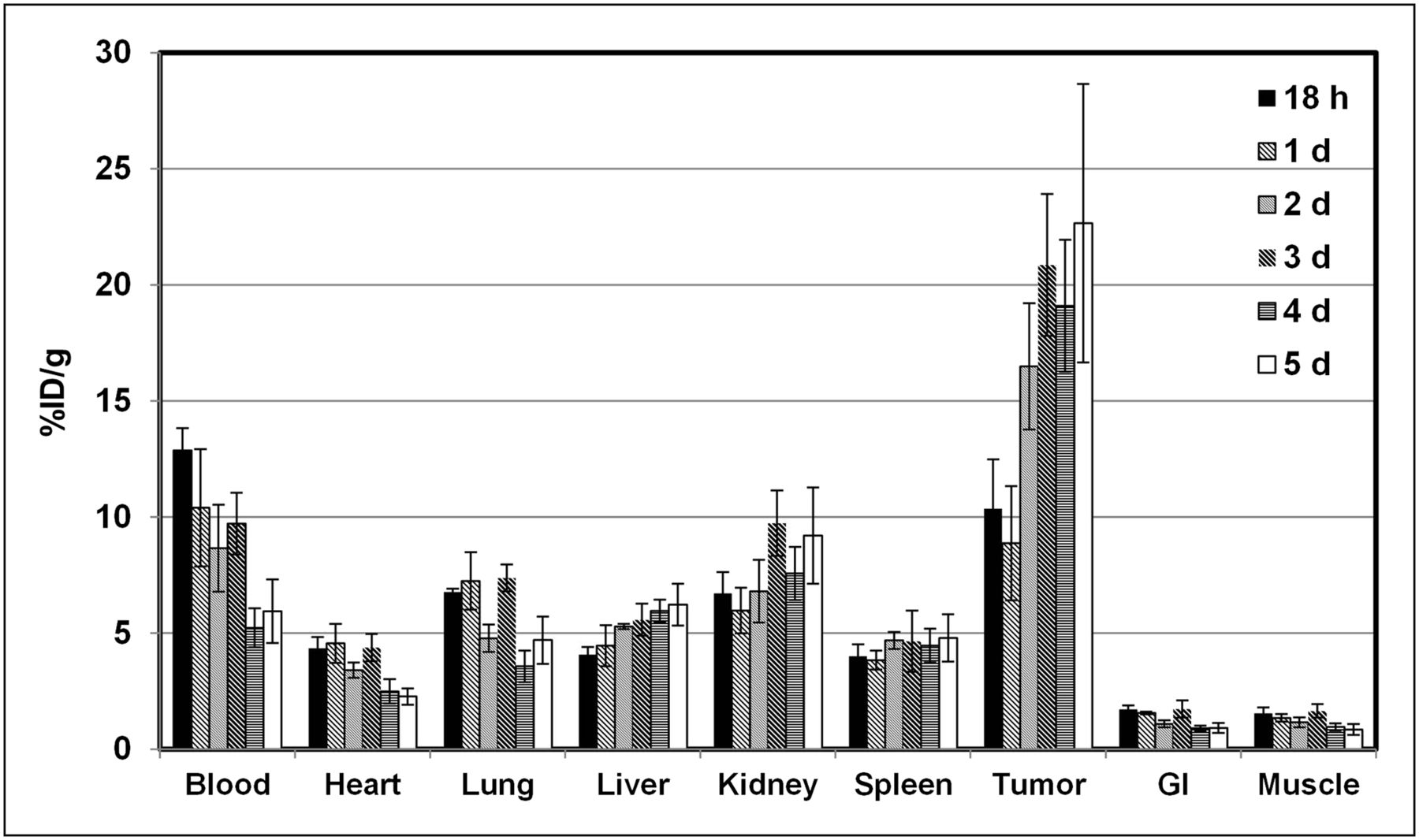
The biodistribution of 89Zr-df-onartuzumab was determined in MKN-45 xenografts from 18 h to 5 d (Fig. 2). 89Zr-df-onartuzumab uptake (%ID/g) in the nontarget tissues—blood, heart, lungs, gastrointestinal tract, and muscle—decreased slowly, with less than a 50% reduction in %ID/g from 18 h to 5 d. In contrast, uptake in the liver and kidneys increased over the time course, suggesting hepatobiliary and renal clearance. At 18 h, 89Zr-df-onartuzumab uptake in tumor was comparable to that in blood, kidneys, and lung, but from 2 to 5 d, uptake in tumor was at least 2-fold greater than that in blood, kidneys, and lung. At 2, 3, 4, and 5 d, the tumors had the highest uptake, ranging from 16.5 to 22.5 %ID/g. In a similar biodistribution study using U87-MG xenografts, the uptake in nontarget tissues was comparable to the results of the MKN-45 xenograft study shown in Figure 2. However, uptake in the U87 tumors was 1.8- and 3.0-fold lower than that in the MKN-45 tumors at 3 and 5 d, respectively. The highest TM ratios of 13.2, 26.5, and 26.6 occurred at 2, 3, and 5 d, respectively, for the MKN-45 xenografts, whereas TM ratios for the U87 xenografts ranged from 7:1 to 9:1 over the 5-d time course (Fig. 3). The 2- to 3-fold reduction in uptake by U87 tumors is consistent with the lower Met expression of this cell line.

Biodistribution of 89Zr-df-onartuzumab in MKN-45 xenografts from 18 h to 5 d. Each bar represents %ID/g ± SD of 89Zr-df-onartuzumab (n = 5, 18-h group; n = 4, other time points). GI = gastrointestinal tract.

Comparison of TM ratios (%ID/g) of 89Zr-df-onartuzumab in MKN-45 and U87-MG xenografts from 18 h to 5 d. Bars represent mean TM ± SD (n = 4). Bars at 3 d include TM from 89Zr-df-onartuzumab competitive blocking study in MKN-45 xenografts injected with 89Zr-df-onartuzumab only or 89Zr-df-onartuzumab plus 10 nmol onartuzumab.
Blocking studies were performed with MKN-45 xenografts in which 89Zr-df-onartuzumab (0.0133 nmol onartuzumab) or 89Zr-df-onartuzumab plus 10 nmol of unlabeled onartuzumab was injected. Uptake (%ID/g) determined after 3 d was comparable for both groups in the blood, heart, lungs, spleen, liver, gastrointestinal tract, and muscle. Kidney uptake increased approximately 2.5-fold, and tumor uptake decreased approximately 3.8-fold, suggesting competitive blocking by unlabeled onartuzumab in the tumor. The TM ratios for the blocked group, compared with mice injected with 89Zr-df-onartuzumab alone, were significantly reduced (67%, P < 0.05), indicating specific Met binding in vivo (Fig. 3).
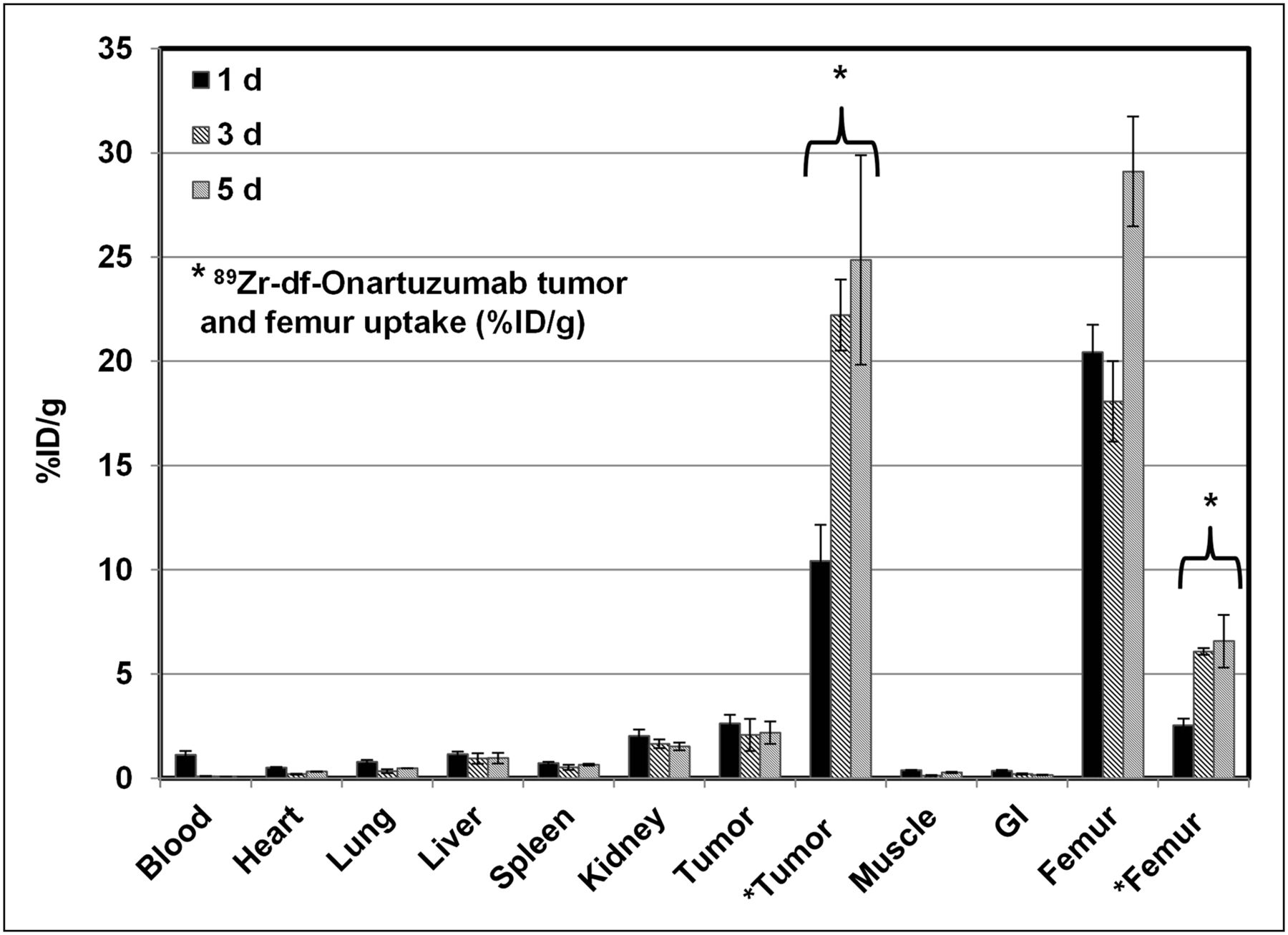
To determine the biologic fate of free 89Zr that may result from 89Zr-df-onartuzumab metabolism, the biodistribution of 89Zr-oxalate (which has a biodistribution characteristic of zirconium (22)) was determined in MKN-45 xenograft mice from 1 to 5 d (Fig. 4). The highest retention and accumulation of radioactivity, ranging from 20 to 30 %ID/g, occurred in bone over the entire course and was 3- to 8-fold higher than 89Zr-df-onartuzumab bone uptake. The greatest clearance of radioactivity from 1 to 5 d was observed in blood (>90%), whereas clearance from other tissues was slower (55%–10% decreases). The tumors retained at least 80% of the radioactivity from 1 to 5 d, with uptake ranging from 2.1 to 2.6 %ID/g, which was higher than in any other tissues except bone. 89Zr tumor uptake (2 %ID/g) represented less than 10% of the 89Zr-df-onartuzumab tumor uptake (22 and 25 %ID/g) at 3 and 5 d, respectively.

Biodistribution of 89Zr-oxalate in MKN-45 xenografts after 1, 3, and 5 d; 89Zr-df-onartuzumab tumor and femur uptake is used for comparison. Each bar represents %ID/g ± SD (89Zr-oxalate, n = 3; 89Zr-df-onartuzumab, n = 3). GI = gastrointestinal tract.
The uptake (%ID/g) of 89Zr-df-onartuzumab and 76Br-onartuzumab, in blood, muscle, and MKN-45 tumors from 6 to 120 h, is compared in Figure 5. 89Zr-df-onartuzumab tumor uptake remained steady or increased over the 5-d time course, whereas 76Br-onartuzumab tumor uptake steadily decreased over the 3-d time course (Fig. 5). Although muscle uptake over time was similar for both tracers, the radioactivity remaining in the blood differed for each agent. After 2 d, 89Zr-df-onartuzumab tumor uptake exceeded the blood levels, whereas 76Br-onartuzumab tumor uptake mirrored blood levels over the entire time course. Thus, 76Br-onartuzumab showed lower retention in the tumors relative to 89Zr-df-onartuzumab, with clearance similar to the radioactivity in blood.
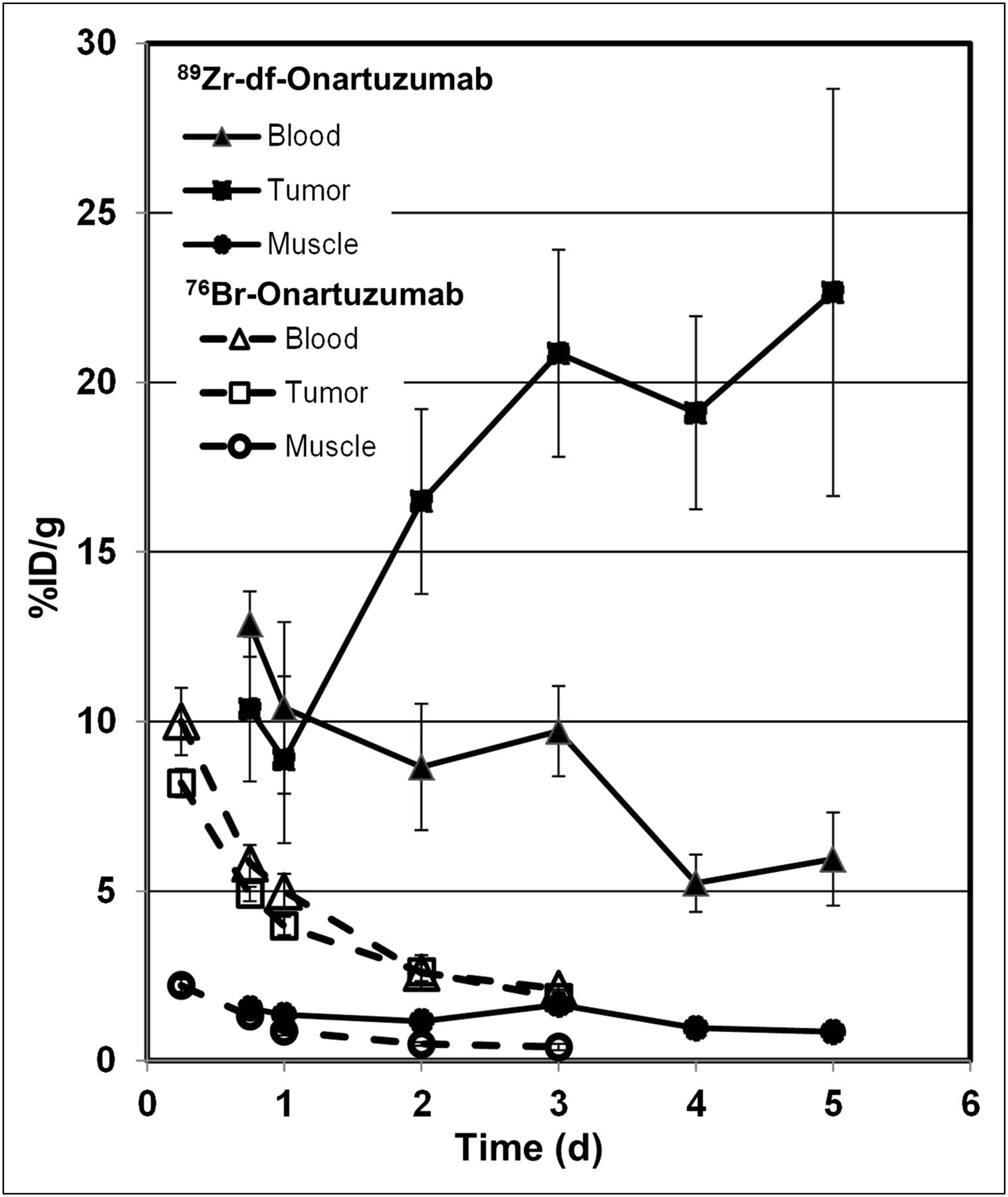
Comparison of blood, tumor, and muscle uptake of 89Zr-df-onartuzumab and 76Br-onartuzumab in MKN-45 xenografts from 6 h to 5 d. Each bar represents %ID/g ± SD of 89Zr-df-onartuzumab (n = 5 [18 h]; n = 4 [other times]) or 76Br-onartuzumab (n = 4).
89Zr-df-onartuzumab had the highest TM ratios—19.6 and 26.6—at 96 and 120 h, respectively (Table 1). Over the entire time course, TM ratios for 76Br-onartuzumab were approximately 2-fold lower than those for 89Zr-df-onartuzumab (Table 1). The 76Br-onartuzumab TM ratios remained relatively unchanged from 24 to 72 h, whereas the 89Zr-df-onartuzumab TM ratios increased from 24 to 120 h (Table 1). Similarly, 76Br-onartuzumab tumor-to-blood ratios were always less than 1 at 6, 16, 24, 48, and 72 h, whereas 89Zr-df-onartuzumab TM ratios were approximately 2 at 72 h and increased to approximately 4 at 120 h.
Small-Animal PET Studies
Small-animal PET studies with 76Br-onartuzumab were performed in MKN-45 or U87-MG xenografts, with imaging from 18 h to 3 d. MKN-45 tumors were visualized as early as 18 h, with image quality improving at 24 h and attaining a TM ratio of approximately 5 (Fig. 6A). Despite an increase the 76Br-onartuzumab dose from 11.1 to 18.5 MBq, images obtained at 48 and 72 h showed no improvement, because of relatively low levels of radioactivity and lack of 76Br-onartuzumab retention in tumors. Therefore, 24 h proved to be the optimum imaging time. At 24 h, U87-MG tumors were more difficult to discern than MKN-45 tumors, consistent with their lower Met expression level.
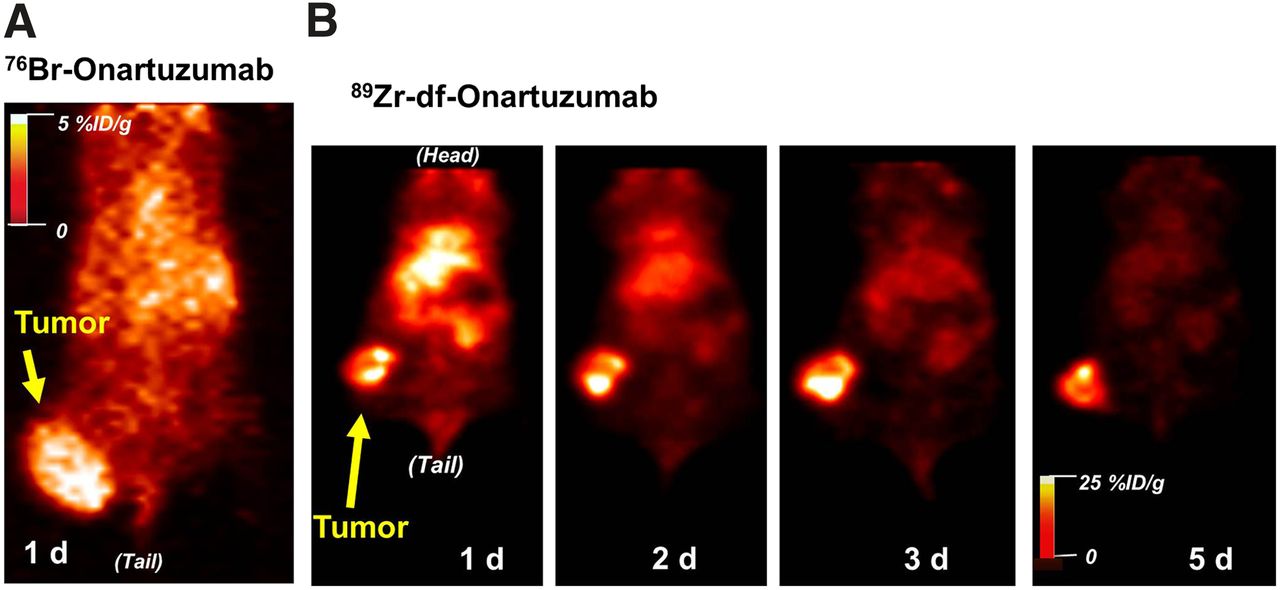
Representative coronal PET images from mouse with MKN-45 tumor on right thigh injected with 76Br-onartuzumab (tumor volume, ∼0.7 cm3) (A) and 89Zr-df-onartuzumab (tumor volume, ∼0.4 cm3) (B).
Imaging studies of MKN-45 xenografts with 89Zr-df-onartuzumab indicated that although tumors could be visualized at 18 h, image quality improved over the 5-d period (Fig. 6B). At 5 d, the MKN-45 TM ratio was 26:1; the increase in TM ratios over the 5-d period reflects better tumor retention and clearance from nontarget tissues.
Correlation of 76Br-Onartuzumab and 89Zr-df-Onartuzumab Uptake with Shed Met and Tumor Met
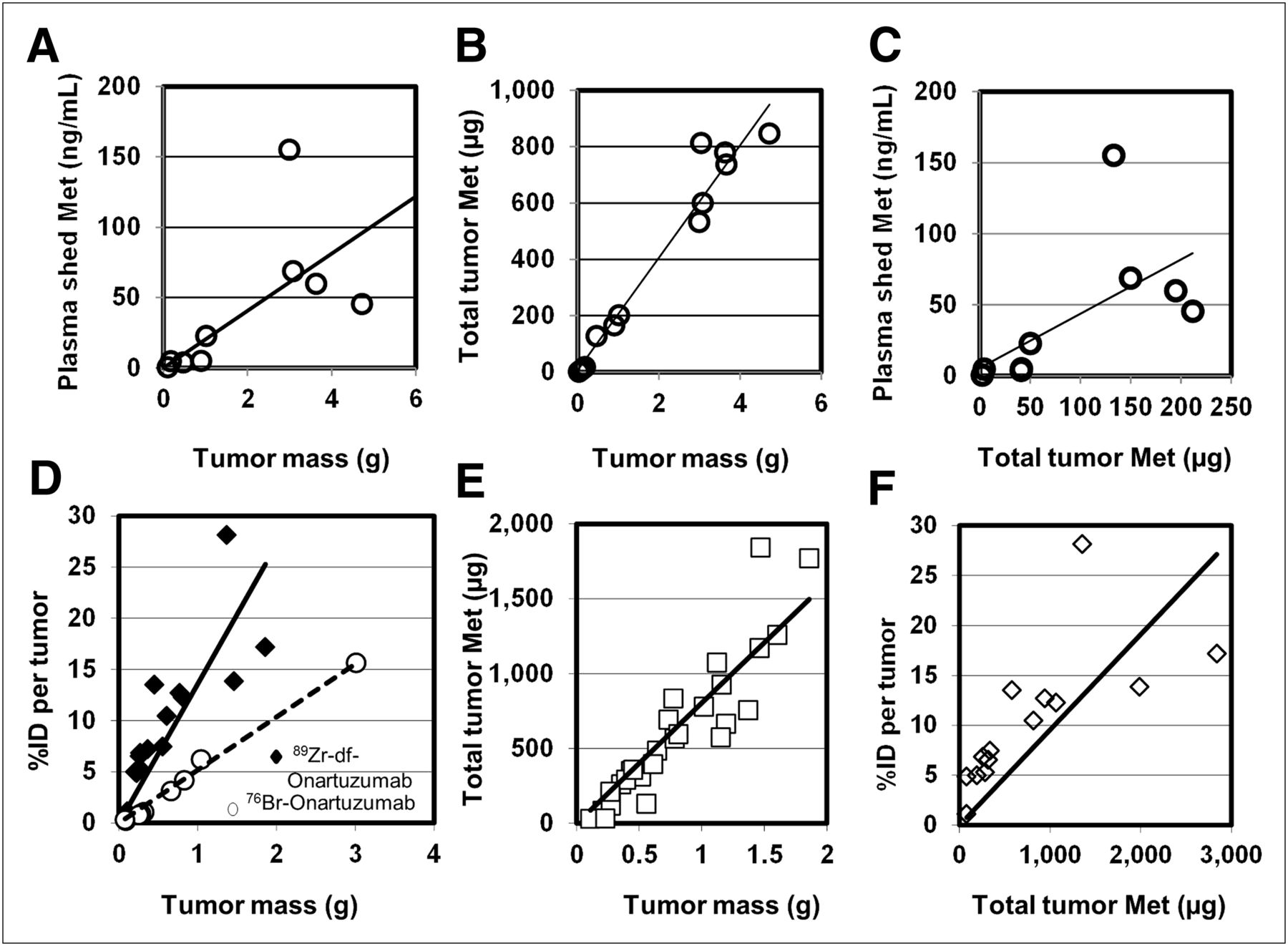
Plasma and tumor samples from naïve MKN-45 xenografts or MKN-45 xenografts injected with 76Br-onartuzumab or 89Zr-df-onartuzumab were analyzed for shed Met ectodomain and total tumor Met content, and then the potential associations between these values to tumor mass or the total tumor radioactive content (percentage injected dose per tumor) were determined (Fig. 7). In naïve xenografts, significant direct correlations were found between tumor mass and shed Met (Fig. 7A), tumor mass and total tumor Met content (Fig. 7B), and total tumor Met content and shed Met (Fig. 7C). The total Met contents of tumor samples excised from xenografts injected with 89Zr-df-onartuzumab were also significantly correlated with the tumor mass (Fig. 7E) and with the 89Zr-df-onartuzumab total tumor uptake (%ID) (Fig. 7F). Similar results were obtained using tumor samples from xenografts injected with 76Br-onartuzumab (data not shown). The similarity in correlations from naïve tumors and tumors labeled with 76Br-onartuzumab or 89Zr-df-onartuzumab indicates that the tracer did not interfere with the Met content determinations. The lower slope observed in the correlation of 76Br-onartuzumab tumor uptake to tumor mass, compared with that of 89Zr-df-onartuzumab (Fig. 7D), most likely reflects the overall decreased tumor retention of 76Br-onartuzumab. The significant correlations between 89Zr-df-onartuzumab or 76Br-onartuzumab tumor uptake and total tumor Met content (Fig. 7F) indicate that radiolabeled onartuzumab tumor uptake directly reflects tumor Met concentrations. Plasma shed Met values from xenografts injected with 76Br-onartuzumab or 89Zr-df-onartuzumab also correlated significantly with tumor mass (data not shown; P < 0.0001 and < 0.03, respectively), comparable to results with the naïve xenografts, indicating that neither 76Br-onartuzumab nor 89Zr-df-onartuzumab affected Met shedding.

Correlation of plasma shed Met, total Met tumor content, and radioactive tumor uptake (%ID per tumor) with tumor mass and Met abundance (all correlations determined using Spearman nonparametric analysis). (A–C) Correlations of plasma and tumor samples from naïve MKN-45 xenografts: r = 0.8167, P < 0.02 (A); r = 0.9321, P < 0.0001 (B); and r = 0.8167, P < 0.01 (C). (D–F) Tumor samples from MKN-45 xenografts previously injected with radiolabeled onartuzumab. (D) Correlations of radiolabeled onartuzumab tumor uptake to tumor mass: 76Br-onartuzumab (○), %ID per tumor determined after 24 h (r = 0.9879; P < 0.0001); 89Zr-df-onartuzumab (◆), %ID per tumor determined after 3, 4, and 5 d (r = 0.9357; P < 0.0001). (E) Correlation of total tumor Met to mass: tumor samples were taken after 0.75, 1, 2, 3, 4, and 5 d of 89Zr-df-onartuzumab uptake and assayed for total Met content (r = 0.9721; P < 0.0001). (F) Correlation of 89Zr-df-onartuzumab tumor uptake (%ID per tumor after 3, 4, and 5 d of uptake) to total tumor Met determinations (r = 0.9385; P < 0.0001).
DISCUSSION
Onartuzumab labeled with 76Br or 89Zr retained most of its biologic activity (>80% immunoreactivity) and displayed nanomolar binding affinity for Met. Both agents could quantitatively distinguish a high-Met-expressing cell line (MKN-45) from a low-Met-expressing cell line (U87-MG) in vitro. Although these in vitro parameters were similar, in vivo biodistribution and imaging studies revealed that 89Zr-df-onartuzumab attained higher tumor–to–nontarget tissue ratios over time, providing higher-quality images than 76Br-onartuzumab in MKN-45 xenografts.
Compared with 89Zr-df-onartuzumab, 76Br-onartuzumab did not accumulate in MKN-45 tumors at later time points but decreased at a rate similar to that in nontarget tissues, resulting in no improvement of TM ratios over time. The lack of further accumulation may in part be related to the direct labeling method used for 76Br-onartuzumab; available tyrosine residues are labeled in the presence of a strong oxidizing agent, and excessive protein oxidation can result in lower immunoreactivity. In addition, directly labeled antibodies internalized into cells are characteristically subject to debromination, thereby losing their label (23). In this case, onartuzumab behaves as an antagonist, unable to cause dimerization; thus, internalization would be expected to be minimal and not necessarily mediated by activation of Met (10). Because 76Br-onartuzumab immunoreactivity was preserved in our studies, in vivo debromination and metabolism are most likely responsible for the lower tumor uptake relative to 89Zr-df-onartuzumab. Alternative radiosynthetic methods have been described for labeling antibodies with 76Br, including indirect methods and an enzymatic method (bromoperoxidase), which claim to improve in vivo stability and thereby increase tumor accumulation and retention (7).
In contrast, indirectly labeled antibodies coupled with a metal chelate, such as 89Zr-df-onartuzumab, tend to have increased accumulations in target cells because of trapping of radioactive metabolites from internalization (23). Our results are similar to those of other studies in which tumor uptake was increased with monoclonal antibodies labeled with 89Zr, a residualizing radionuclide, compared with 124I, a nonresidualizing radionuclide (9). In MKN-45 tumors, we observed substantially greater retention of 89Zr-df-onartuzumab (22 and 25 %ID/g at 3 and 5 d, respectively) than of 89Zr-oxalate (2.1 and 2.2 %ID/g at 3 and 5 d, respectively), indicating that most of the tumor uptake would be expected to represent binding to Met rather than free 89Zr.
Although 89Zr-df-onartuzumab uses an indirect labeling method conferring increased tumor retention, we observed a high bone uptake ranging between 6 and 7 %ID/g at 3 and 5 d, respectively, that could theoretically affect clinical application. In agreement with our results, 89Zr-oxalate and other solvated or chelated forms of 89Zr had high bone accumulation in mice, and this accumulation was primarily associated with the bones and epiphyses rather than bone marrow (22). Further, Abou et al. (22) showed that the bone uptake was dependent on the strength of the chelate. Although these preclinical mouse studies predicted potentially high bone uptake of free 89Zr in human studies, successful 89Zr immuno-PET clinical studies have been performed in which bone uptake and radiation doses to bone marrow were minimal (24), suggesting that with the appropriate radiochemistry and a strong chelate, high 89Zr bone uptake can be avoided.
In addition to the longer radioactive half-life, which better accommodates the longer biologic half-life of onartuzumab and increased tumor retention, 89Zr-df-onartuzumab in vivo tumor uptake was Met-specific and sensitive to changes in Met densities. Unlabeled onartuzumab significantly blocked tumor uptake, indicating that 89Zr-df-onartuzumab and onartuzumab are competing for the same binding site. The substantially decreased 89Zr-df-onartuzumab uptake observed in U87-MG tumors relative to MKN-45 tumors represents reduced Met densities in U87-MG tumors, which are known to have low Met expression. Further, the significant correlations among 89Zr-df-onartuzumab tumor uptake, tumor mass, and total tumor Met content confirm that 89Zr-df-onartuzumab tumor uptake is directly related to tumor Met density. However, in larger tumors 89Zr-df-onartuzumab uptake may be decreased because of necrosis rather than real changes in Met densities of the viable tumor tissue. In this case, for the 89Zr-df-onartuzumab uptake to accurately correspond to the tumor Met density, viable uptake excluding the necrotic regions would need to be determined from the PET images. The presence of shed Met did not appear to alter the 76Br-onartuzumab biodistributions in nude mice versus MKN-45 xenografts, which is most likely a consequence of the high molar ratio of 89Zr-df-onartuzumab to shed Met (>100-fold). Although shed Met concentrations were comparable from naïve and radiolabeled onartuzumab, suggesting that radiolabeled onartuzumab does not interact with shed Met, this does not preclude the possibility that radiolabeled onartuzumab binds to a distinct site that does not cross-react with the immunoassay detection antibody. More importantly, these results indicate that shed Met would not be expected to interfere with the delivery of radiolabeled onartuzumab to its target.
CONCLUSION
The results of our study indicate that 89Zr-df-onartuzumab may prove to be an effective immuno-PET agent in humans. However, onartuzumab does not recognize murine Met, and thus our preclinical studies do not address onartuzumab uptake that might occur in human tissues that normally express Met. Initial imaging studies in humans with 89Zr-df-onartuzumab designed to determine uptake in nontarget tissues will be important for determining imaging feasibility. Because 89Zr-df-onartuzumab represents the radiolabeled version of the therapeutic onartuzumab, this agent may aid in drug development by identifying tissues that may be subject to onartuzumab-associated toxicities, assessing responses to onartuzumab treatment, establishing appropriate dosing for optimal tumor targeting, and tracking in vivo the disposition of onartuzumab in individual patients. 89Zr-df-onartuzumab imaging may also prove useful in the development of small-molecule Met tyrosine kinase inhibitors and other Met-targeted agents for monitoring therapeutic responses in which decreased uptake would be expected in Met-positive tumors. More generally, 89Zr-df-onartuzumab has potential utility for imaging Met to identify patients for treatment with Met-targeted therapeutics and to identify the emergence of Met-driven acquired resistance to other molecularly targeted cancer therapies.
DISCLOSURE STATEMENT
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
No potential conflict of interest relevant to this article was reported.
Footnotes
Published online Aug. 23, 2012.
- © 2012 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication December 26, 2011.
- Accepted for publication May 21, 2012.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Immuno-PET Detects Changes in Multi-RTK Tumor Cell Expression Levels in Response to Targeted Kinase Inhibition
- Light-Induced Radiosynthesis of 89Zr-DFO-Azepin-Onartuzumab for Imaging the Hepatocyte Growth Factor Receptor
- PET Imaging of Receptor Tyrosine Kinases in Cancer
- c-Met PET Imaging Detects Early-Stage Locoregional Recurrence of Basal-Like Breast Cancer
- Emerging Opportunities for c-MET Visualization in the Clinic
- PET of c-Met in Cancer with 64Cu-Labeled Hepatocyte Growth Factor
- Anti-MET ImmunoPET for Non-Small Cell Lung Cancer Using Novel Fully Human Antibody Fragments
- In Vivo Visualization of MET Tumor Expression and Anticalin Biodistribution with the MET-Specific Anticalin 89Zr-PRS-110 PET Tracer
- Interrogating Tumor Metabolism and Tumor Microenvironments Using Molecular Positron Emission Tomography Imaging. Theranostic Approaches to Improve Therapeutics