


Abstract
Our objective was to measure (R)-11C-PK11195 binding as an indirect marker of neuronal damage after traumatic brain injury (TBI). Methods: Dynamic (R)-11C-PK11195 PET scans were acquired for 8 patients 6 mo after TBI and for 7 age-matched healthy controls. (R)-11C-PK11195 binding was assessed using the simplified reference tissue model. Because of widespread traumatic changes in TBI, an anatomic reference region could not be defined. Therefore, supervised cluster analysis was used to generate an appropriate reference tissue input. Results: Increased whole-brain binding of (R)-11C-PK11195 was observed in TBI patients. Regional analysis indicated that increased (R)-11C-PK11195 binding was widespread over the brain. Conclusion: Six months after TBI, there was a prolonged and widespread increase in (R)-11C-PK11195 binding, which is indicative of diffuse neuronal damage.
Traumatic brain injury (TBI) is the leading cause of death and disability among young people under the age of 40 y. Frequently, survivors of TBI (and their relatives) have to deal with the devastating consequences of prolonged neurologic and cognitive impairments (1). Worldwide, an estimated 1.2 million people are killed in road crashes each year and as many as 50 million are injured. According to the World Health Organization, these figures will increase by about 65% over the next 20 y (2). From this point of view, TBI is a tremendous burden on society.
In TBI, structural intracranial abnormalities such as hemorrhages and edema can be visualized by CT and MRI. Complementary to those, PET techniques can be used to demonstrate and evaluate posttraumatic disturbances in, for example, brain metabolism or brain function. Already by the mid 1980s, PET studies on TBI patients had been performed, mainly with 18F-FDG. Langfitt et al. found extensive disturbances in glucose metabolism beyond structural abnormalities demonstrated by CT and MRI (3). Their findings were in line with studies performed by Rao et al. (4). They found that the results from PET studies closely corresponded to the site and extent of cerebral dysfunction, determined by neurologic and behavioral examinations, whereas CT did not. Follow-up CT studies, however, demonstrated structural abnormalities consistent with the early PET findings, supporting the predictive value of 18F-FDG PET (4). More recently, diffuse cerebral hypometabolism has also been demonstrated in boxers with neurologic and cognitive disabilities as a consequence of chronic TBI and in war veterans with persistent postconcussive symptoms (5,6).
Today, research in the field of TBI is becoming increasingly focused on posttraumatic neuroinflammation. It has been suggested that microglial activation may play an important role in a variety of neuroinflammatory and neurodegenerative diseases (7,8). Microglial activation is thought to be closely associated with neuronal damage. As such, activated microglia may also be involved after head trauma. In addition, microglia are capable of synthesizing immune mediators, including cytokines, chemokines, and complement activation proteins, and of expressing specific receptors of these immune mediators (9). In this context, the putative role of activated microglia in TBI is that they serve as the major antigen-presenting cells in brain tissue, which are crucial in the highly complex neuroinflammatory cascade after brain injury. Microglial imaging in TBI patients can be of meaningful clinical and diagnostic value in terms of visualization and quantification of active disease processes, though the long-term effects of microglial activation in the posttraumatic neuropathologic sequelae after TBI, together with its implications for neurologic outcome, have not been elucidated yet.
(R)-PK11195 (1-[2-chlorophenyl]-N-methyl-N-[1-methyl-propyl]-3-isoquinoline carboxamide) is a selective ligand for activated microglia. When labeled with the positron emitter 11C, it can be used to quantify microglial activation in vivo using PET. This approach has already been used in the study of several neurodegenerative disorders (7,10,11). The main purpose of the present study was to quantify (R)-11C-PK11195 binding in patients with moderate and severe TBI 6 mo after injury.
MATERIALS AND METHODS
Subjects
Eight patients (5 men and 3 women; age range, 18–63 y) with moderate (n = 5) or severe (n = 3) TBI (total Glasgow Coma Scale on the day of trauma [GCS], 9–13 and ≤8, respectively) were studied (Table 1). In addition, 7 age- and sex-matched healthy controls (4 men and 3 women; age range, 18–59 y) were included after a standardized physical and mental health screening, including a normal brain MRI result as judged by a neuroradiologist. Written informed consent was obtained from all patients or their next of kin and from all healthy controls. In accordance with the Declaration of Helsinki, the clinical research protocol had been approved by the Medical Ethics Review Committee of the VU University Medical Center, Amsterdam, The Netherlands.
Patient Characteristics
Study Design
For all subjects, both (R)-11C-PK11195 and MRI scans of the brain were acquired within the same week. In TBI patients, the procedure was performed 6 mo after injury. None of the TBI patients or the healthy controls was on nonsteroidal antiinflammatory drugs or benzodiazepines.
(R)-11C-PK11195 PET Scans
(R)-11C-PK11195 PET scans were acquired using an ECAT EXACT HR+ scanner (Siemens/CTI), equipped with a neurologic insert. (R)-11C-PK11195 was produced according to the method of Shah et al. (12). After a 10-min transmission scan in 2-dimensional acquisition mode, a 3-dimensional dynamic (R)-11C-PK11195 scan was obtained, consisting of 23 frames of increasing length and a total acquisition time of 62.5 min. A bolus of (R)-11C-PK11195 was injected intravenously using a fully automated infusion pump. Administered (R)-11C-PK11195 activity was not significantly different between patients (422 ± 49 MBq) and healthy controls (438 ± 37 MBq; P = 0.67), nor was there a significant difference in specific activity between patients (48.1 ± 16.5 GBq/μmol) and healthy controls (75.1 ± 30.3 GBq/μmol; P = 0.065). Injected mass was 3.3 ± 0.9 μg for TBI patients and 1.5 ± 0.3 μg for healthy controls (P = 0.003). All PET scans were reconstructed using a Fourier rebinning algorithm plus 2-dimensional filtered backprojection, with a Hanning filter at a cutoff of 0.5 times the Nyquist frequency, resulting in an image resolution of approximately 7 mm in full width at half maximum (13). All data were corrected for decay, dead time, randoms, scatter, and tissue attenuation. A zoom factor of 2 and an image matrix size of 256 × 256 × 63 were used, resulting in a voxel size of 1.2 × 1.2 × 2.4 mm.
Kinetic and Parametric Analysis
Parametric (R)-11C-PK11195 nondisplaceable binding potential (BPND) images were generated using reference parametric mapping (14), a basis function implementation of the simplified reference tissue model (15). A modified supervised cluster analysis algorithm (SVCA4) was used to extract the reference tissue input curve (16,17). In a previous study, the use of a simplified reference tissue model for analyzing (R)-11C-PK11195 data 6 mo after TBI was validated (18).
Volume-of-Interest Definition
PET and MRI scans were coregistered using the software package MIRIT (19). Volumes of interest for cerebellum, frontal lobe, parietal lobe, temporal lobe, occipital lobe, ventromedial prefrontal cortex, hippocampus, cingulate gyrus, caudate nucleus, basal ganglia, midbrain, pons, and medulla, including posttraumatic intracranial lesions as determined by CT and MRI, were defined manually on the coregistered MRI scans using a dedicated 3-dimensional software package (http://www.idoimaging.com/program/172). Regional values of BPND were obtained by projecting all volumes of interest onto the parametric (R)-11C-PK11195 BPND images. Whole-brain BPND was defined as the volume-weighted average of these regional volume-of-interest values.
Clinical Evaluation
Six months after injury, the extended Glasgow Outcome Scale (GOS-E) was defined in all TBI patients (1 = dead, 2 = vegetative state, 3 = lower severe disability, 4 = upper severe disability, 5 = lower moderate disability, 6 = upper moderate disability, 7 = lower good recovery, and 8 = upper good recovery). Patient characteristics are shown in Table 1.
Statistical Analysis
The Student t test was used to assess differences in brain BPND between TBI patients 6 mo after head injury and age- and sex-matched healthy controls. Significance level was defined as P < 0.05.
RESULTS
Subjects
Patient characteristics are shown in Table 1. The mean age of the 8 consecutive TBI patients was 40.5 ± 17.0 y (range, 18–63 y). The mean initial GCS was 9 (range, 3–13). GOS-E at 6 mo ranged from 4 to 8.
(R)-11C-PK11195 PET
Whole-brain analysis revealed a significantly (P = 0.02) increased (R)-11C-PK11195 BPND in TBI patients (BPND = 0.22 ± 0.08) 6 mo after head injury, as compared with age-and sex-matched healthy controls (BPND = 0.12 ± 0.06) as illustrated in Figure 1. Increased (R)-11C-PK11195 BPND was found not only in the affected brain regions but also in nontraumatic brain regions as determined by MRI (Fig. 2). On an individual basis, no clear correlation was found between the severity of head injury in terms of GCS or outcome (GOS-E) and whole brain (R)-11C-PK11195 BPND. Maximum (R)-11C-PK11195 BPND was found in the thalamus in 6 of 8 TBI patients. In 1 severe TBI patient (GCS 3) and 1 moderate TBI patient (GCS 13), the maximum (R)-11C-PK11195 BPND was found in the hippocampus and medulla, respectively (Table 1). Increased (R)-11C-PK11195 BPND was found in several brain areas of TBI patients: left and right frontal lobe, left and right thalamus, left parietal lobe, right temporal lobe, hippocampus and putamen, midbrain and pons (BPND = 0.36 ± 0.12, P < 0.01) (Table 2). The higher injected mass in patients cannot explain the higher BPND. If anything, it should have resulted in lower BPND values.
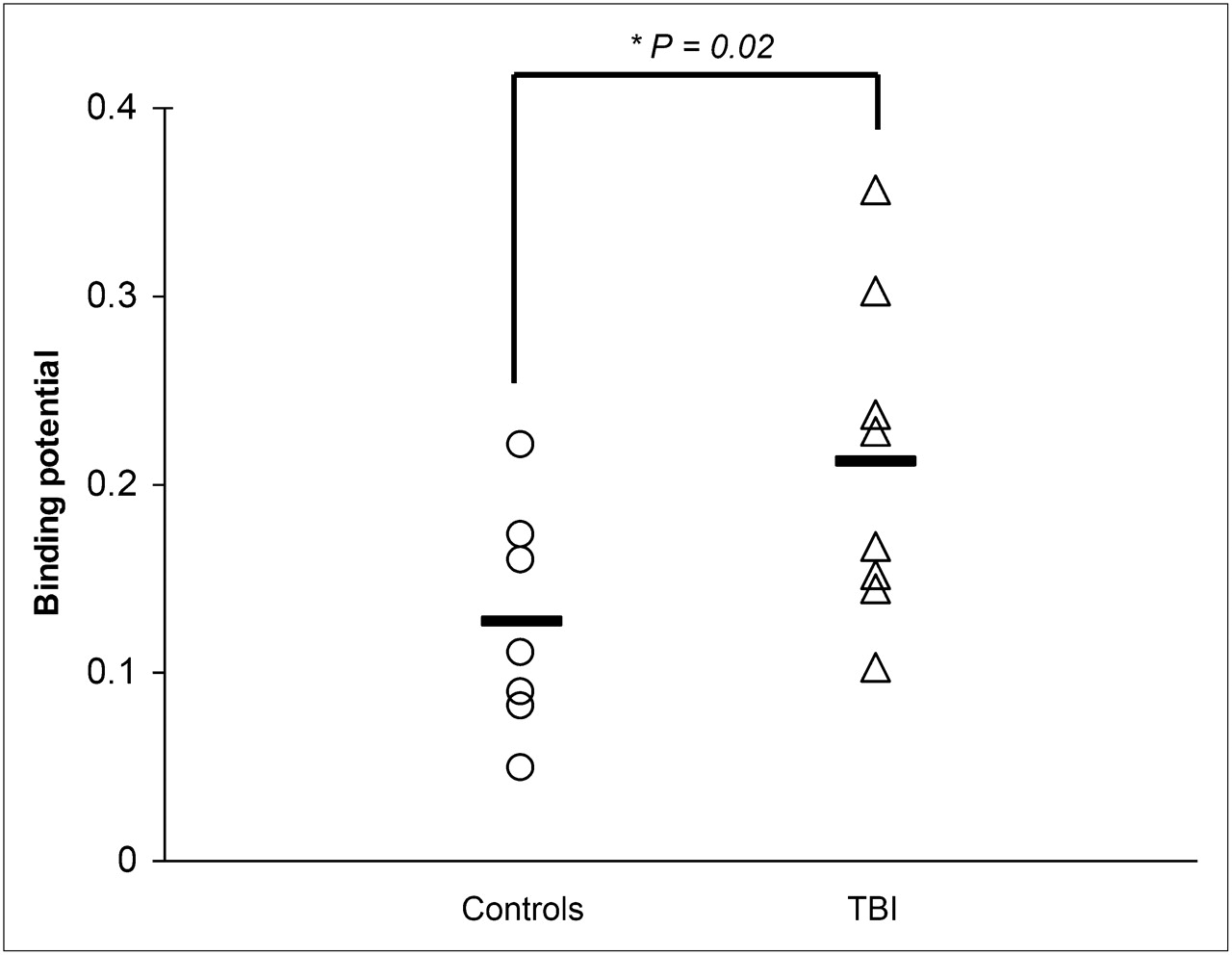
Scatterplot of whole brain (R)-11C-PK11195 BPND in healthy controls and TBI patients 6 mo after injury. Mean values are represented by horizontal lines.

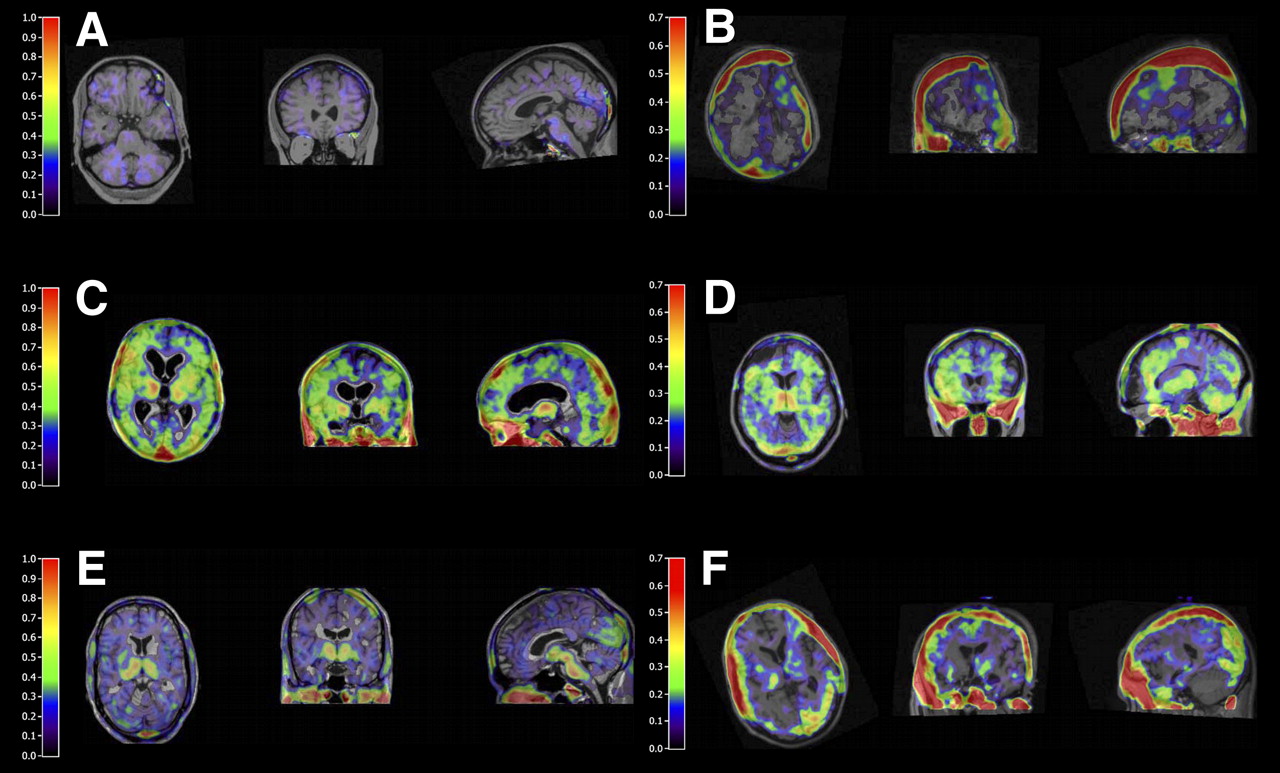
Parametric (R)-11C-PK11195 BPND images for healthy control (A), TBI patients with focal injury (B, D, and F), and TBI patients with diffuse injury (C and E). Images were generated using reference parametric mapping, with reference tissue input function generated by supervised cluster analysis. (A) Representative picture of healthy control. (B) Increased ipsilateral (R)-11C-PK11195 BPND at site of hemicraniectomy. (C) Significant bilateral globally increased (R)-11C-PK11195 BPND. (D) Increased pericontusional thalamic and cerebellar (R)-11C-PK11195 BPND. (E) Increased thalamic (R)-11C-PK11195 BPND at midbrain and occipital lobe. (F) Contrecoup increased (R)-11C-PK11195 BPND in parietooccipital lobe.
(R)-11C-PK11195 BPND for Specific Brain Regions in TBI Patients and Healthy Controls
DISCUSSION
To our knowledge, this was the first proof-of-concept study that showed a significant increase in both whole-brain and regional (R)-11C-PK11195 BPND 6 mo after TBI, indicating prolonged and widespread microglial activation after head injury.
The role of activated microglia after TBI is still poorly understood. In vitro studies, animal models, and in vivo clinical studies have demonstrated that activated microglia produce a myriad of inflammatory mediators (7,8,20). Apart from being an important defense mechanism against invading pathogens, activated microglia may also be involved in the pathogenesis of neuronal brain damage. In TBI, as a consequence of the initial insult, dying or damaged neurons activate resting microglia in brain tissue. Subsequently, these activated microglia produce not only neurotrophic but also neurotoxic factors, which may further damage surrounding neurons, resulting in ongoing bystander neurotoxicity (21). As clearly stated by Banati, injured brain is less static than commonly thought and shows subtle glial responses even in regions that appear macroanatomically stable. Glial activation is not solely a sign of tissue destruction but may also indicate disease-induced adaptation or plasticity (22).
TBI can be seen as a global brain disease. Widespread pathophysiologic changes without any structural damage of affected regions, combined with the generally low signal-to-noise ratio of (R)-11C-PK11195, preclude an anatomic definition of a reference region (e.g., cerebellum). In supervised cluster analysis, by using predefined kinetic classes, a gray matter reference tissue input without specific tracer binding (i.e., without activated microglia) is extracted (17). Using SVCA4, a specific implementation of supervised cluster analysis, it was possible to generate an appropriate and reliable reference tissue input and analyze the data with the validated reference tissue approach.
From this series, increased and prolonged (R)-11C-PK11195 binding was found in TBI patients, as compared with healthy controls (Fig. 1). Not only in the ipsilateral but also in the contralateral hemisphere was increased (R)-11C-PK11195 binding demonstrated, indicating more widespread neuronal damage (Fig. 2). Clinical reading of MRI scans showed far less affected regions in the brains of TBI patients, indicating that (R)-11C-PK11195 may be a sensitive marker for neuronal damage after TBI.
Prolonged microglial activation in specific areas in the ipsi- and contralateral hemispheres stresses the importance of prevention and treatment of secondary injury in management of severe TBI patients. Therefore, 11C-(R)-PK11195 PET could become an attractive additional technique—which needs to be further evaluated—in detecting secondary injury after TBI. Currently, PET in TBI is of limited clinical applicability because of the high costs, complex logistic and infrastructural setup, and time-consuming analysis of limited clinical use.
CONCLUSION
From a scientific point of view, 11C-(R)-PK11195 PET has a high potential to become a sophisticated diagnostic molecular imaging technique that could contribute to the growing knowledge of secondary injury mechanisms in TBI. Further research is needed to elucidate whether and how the prolonged increase in (R)-11C-PK11195 binding after TBI reflects ongoing and possibly preventable progressive neuronal damage. If this is the case, controlling microglial activation may lead to a better outcome after TBI.
DISCLOSURE STATEMENT
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We acknowledge Wiesje van der Flier for statistical support. This study was supported by the Dutch Brain Foundation (9F01.21) and by the Netherlands Organisation for Scientific Research (NWO, VIDI grant 016.066.309). No other potential conflict of interest relevant to this article was reported.
- © 2011 by Society of Nuclear Medicine
REFERENCES
- Received for publication October 8, 2010.
- Accepted for publication February 28, 2011.





{kind=link}
{kind=link}