


Abstract
Cyclooxygenase (COX)-1 and -2 are prostanoid-synthesizing enzymes that play important roles in the regulation of neuroinflammation and in the development of neurodegenerative disorders. However, the specific functions of these isoforms are still unclear. We recently developed 11C-labeled ketoprofen methyl ester as a PET probe that targets the COXs for imaging neuroinflammation, though its responsible isoform is yet to be determined. In the present study, we performed ex vivo and in vivo imaging studies with 11C-ketoprofen methyl ester and determined the contributions of the COX isoforms during the neuroinflammatory process. Methods: To identify the COX isoform responsible for 11C-ketoprofen methyl ester in the brain, we examined the ex vivo autoradiography of 11C-ketoprofen methyl ester using COX-deficient mice. Time-dependent changes in accumulation of 11C-ketoprofen methyl ester during the neuroinflammatory process were evaluated by PET in rats with hemispheric neuroinflammation induced by intrastriatal injection of lipopolysaccharide or quinolinic acid. In both rat models, cell-type specificity of COX isoform expression during neuroinflammation was identified immunohistochemically. Results: Ex vivo autoradiographic analysis of COX-deficient mice revealed a significant reduction of 11C-ketoprofen methyl ester accumulation only in COX-1–deficient mice, not COX-2–deficient mice. PET of rats after intrastriatal injection of lipopolysaccharide showed a significant increase in accumulation of 11C-ketoprofen methyl ester in the inflamed area. This increase was evident at the early phase of 6 h, peaked at day 1, and then returned to basal levels by day 7. In addition, immunohistochemical analysis revealed that the population of activated microglia and macrophages was elevated at the early phase with COX-1 expression but not COX-2. A significant increase in 11C-ketoprofen methyl ester accumulation was also observed at day 1 after intrastriatal injection of quinolinic acid, with increased COX-1–expressing activated microglia and macrophages. Conclusion: We have identified 11C-ketoprofen methyl ester as a COX-1–selective PET probe, and using this, we have also demonstrated a time-dependent expression of COX-1 in activated microglia and macrophages during the neuroinflammatory process in the living brain. Thus, COX-1 may play a crucial role in the pathology of neuroinflammation and might be a critical target for the diagnosis and therapy of neurodegenerative disorders.
Neuroinflammation is an important mechanism in defense responses to pathogenic events but is also recognized as a key component in the progression of neurodegenerative diseases such as Alzheimer disease, Parkinson disease, amyotrophic lateral sclerosis, multiple sclerosis, traumatic brain injury, and HIV encephalitis (1,2). Cyclooxygenase (COX) is a key enzyme in the rate-limiting conversion of arachidonic acid to prostaglandins, one of the proinflammatory factors, and is known to play a crucial role in the inflammatory cascade (3,4). Two distinct COX isoforms, COX-1 and COX-2, have been characterized. COX-1 is classically considered a constitutively expressed housekeeping enzyme and COX-2 an inflammatory inducible enzyme (5,6). In the central nervous system, overexpression of COX-1 and COX-2 in patients with ischemic, traumatic brain injury and in several neurodegenerative diseases such as Alzheimer disease, Parkinson disease, and amyotrophic lateral sclerosis has been reported, indicating that COX-mediated neuroinflammation is a critical component in neuronal degeneration (7–11).
A noninvasive in vivo imaging method to monitor COX expression by PET could be a valuable tool for investigating the role of the COX isoforms in the pathologic processes of neurologic disorders (12). COX inhibitors such as nonsteroidal antiinflammatory drugs labeled with 18F or 11C have been evaluated as PET probes for COX imaging in inflammation. Previously, PET probes that are highly selective to COX-2, such as 18F-desbromo-Dup-697 and 11C-celecoxib, had been developed (13,14). However, the selectivity of these probes to COX-2 proved to be unsatisfactory in vivo when examined by PET. Recently, we developed the use of 11C-ketoprofen methyl ester (the acidic form specific to COX-1; 50% inhibition concentrations, 0.047 μM for COX-1 and 2.9 μM for COX-2) together with a method for efficient, rapid 11C-labeling of 2-arylpropionic acids and their esters (a well-known class of nonsteroidal antiinflammatory drugs) as a PET probe for COX imaging of neuroinflammation (15). A significant and specific accumulation in the inflamed region of rat brain caused by intracranial lipopolysaccharide injection was shown by 11C-ketoprofen methyl ester but not 11C-ketoprofen because of blood–brain barrier permeability (15).
In the present study, we performed PET with 11C-ketoprofen methyl ester to examine time-dependent changes in the contribution of the COX isoforms during the neuroinflammatory processes of 2 different types of pathologic neuronal damage induced by intrastriatal injection of lipopolysaccharide and quinolinic acid. Lipopolysaccharide is a cell-wall component of gram-negative bacteria that is widely used for activating the innate immune system and is reported to cause neuroinflammation with microglial activation (16,17). Quinolinic acid is an excitotoxic N-methyl-D-aspartate receptor agonist reported to induce the death of neurons and astrocytes with excessive induction of nitric oxide and nitric oxide–mediated free radicals (18,19). In this study, we also characterized the selectivity and specificity of 11C-ketoprofen methyl ester to COX-1 and COX-2 in vivo using COX-1–deficient (COX-1−/−) and COX-2–deficient (COX-2−/−) mice, respectively, and identified immunohistochemically the cell-type specificity of COX isoform expression.
MATERIALS AND METHODS
Radiochemistry
11C-labeled ketoprofen methyl ester was synthesized using a rapid 11C-methylation method recently developed by our group (15). The specific radioactivity ranged from 19 to 47 GBq/μmol at the end of synthesis. Both the radiochemical and the chemical purities were greater than 99% as determined by high-performance liquid chromatography (COSMOSIL 5C18-AR-II column [Nacalai Tesque, Inc.]; 4.6-mm internal diameter × 100 mm; particle size, 5 μm; mobile phase, 50:50 acetonitrile:water; flow rate, 1 mL/min; ultraviolet detection, 254 nm; retention time, 5 min).
11C-labeled (R)-N-methyl-N-(1-methylpropyl)-1-(2-chlorophenyl)isoquinoline-3-carboxamide [(R)-PK11195] was synthesized according to the method described by Shah et al. (20), with slight modifications. The specific activity ranged from 44 to 135 GBq/μmol at the end of synthesis. Both the radiochemical and the chemical purities were greater than 99% as determined by high-performance liquid chromatography (COSMOSIL 5C18-AR-II column; 4.6-mm internal diameter × 100 mm; particle size, 5 μm; mobile phase, 65:35 acetonitrile:water; flow rate, 1 mL/min; ultraviolet detection, 215 nm; retention time, 4 min).
Animals
All experimental protocols were approved by the Animal Care and Use Committee of RIKEN Kobe Institute (MAH19-03) and were performed in accordance with the National Institutes of Health principles of laboratory animal care (21). Male mice homozygous-deficient (−/−) for COX-1 and COX-2 (3–4 mo of age) bred on a C57BL/6 × 129/Ola (C57/129) background were obtained from Taconic Farms Inc. These mice and their matching wild-type littermate controls were produced by crossing COX-1 or COX-2 heterozygous mice that had been maintained for more than 35 generations (22,23). Male Sprague–Dawley rats (3 mo old) were obtained from CLEA Japan, Inc. Four to 5 mice were housed in a cage, under a 12-h light–dark cycle (lights off at 20:00) at 23°C ± 1°C and 60% ± 5% humidity and were allowed access to food and water ad libitum.
Generation of Neuroinflammatory Models
Injection of lipopolysaccharide or quinolinic acid into the striatum of rats was performed using a stereotactic apparatus (SR-6N; Narishige Co.) under sodium pentobarbital anesthesia (50 mg/kg). Lipopolysaccharide from Escherichia coli 026:B6 (0.5 μg/μL; Sigma-Aldrich Co.) or quinolinic acid (60 nmol/μL; Sigma-Aldrich Co.) was dissolved in saline and injected into the striatum using a 10-μL syringe with a 26-gauge needle controlled by an automated syringe pump (Muromachi Kikai Co.) at a rate of 0.2 μL/min for 5 min. The coordinates for the stereotactic injection were +0.2 mm anterior/posterior, +3.2 mm lateral, and −5.5 mm dorsal/ventral from the bregma, according to the atlas of Paxinos and Watson (24). After the injection, the needle was kept in the same position for an additional 5 min and then retrieved slowly from the brain.
PET Studies
A venous catheter for PET probe injection was inserted into the tail vein before PET. The rats were anesthetized with a mixture of 1.5% isoflurane and 7:3 nitrous oxide:oxygen and placed on the bed of a small-animal PET scanner (microPET Focus 220; Siemens). The PET scanner has a spatial resolution of 1.4 mm in full width at half maximum at the center of the field of view (220 mm in diameter) and an axial extent at 78 mm. 11C-ketoprofen methyl ester or 11C-PK11195 (∼70 MBq per animal) dissolved in 1 mL of saline was injected via the cannula inserted into the tail vein for 10 s, and emission data were then acquired for 45 min using a 3D list-mode method. The data were reconstructed with standard 2-dimensional filtered backprojection (ramp filter, cutoff frequency at 0.5 cycles per pixel). Regions of interest were placed on the ipsilateral and contralateral sides of the striatum using image processing software (PMOD, version 3.0; PMOD Technologies Ltd.) according to the fused PET/MR T1-weighted image (Supplemental Fig. 1; supplemental materials are available online only at http://jnm.snmjournals.org). Regional uptake of radioactivity in the brain was decay-corrected to the injection time and was expressed as the standardized uptake value (SUV), where SUV = tissue radioactivity concentration (MBq/cm3)/injected radioactivity (MBq) × body weight (g).
Ex Vivo Autoradiography
To assess the quantification of emission data, we performed ex vivo brain slice autoradiography. Because a rapid and transient brain time–activity curve was observed in our preliminary 11C-ketoprofen methyl ester PET study with mice, we decided to use the time point at 15 min to avoid the effect of cerebral blood flow. Fifteen minutes after injection of 11C-ketoprofen methyl ester (∼20 MBq per animal), the mice were sacrificed under deep anesthesia with 3.0% isoflurane and the brain was sliced into coronal sections (1 mm thick) using a brain matrix (RBM-2000C; ASI instruments, Inc.) on ice. The coronal slices were placed on an imaging plate (BAS-SR2040; Fuji Photo Film Co.) for 1 h. The exposed imaging plate was then scanned with a bioimaging analyzer (FLA-7000IR; Fuji Photo Film Co.). To obtain quantitative data from ex vivo autoradiograms, we manually placed regions of interest on the brain regions according to the digital photograph obtained from a photo scanner. Brain regional radioactivity (photostimulable luminescence/mm2) was calibrated to the uptake value (% dose/g), which was defined as 100 × tissue activity concentration (MBq/g)/injected dose (MBq) using whole-brain radioactivity measured by a γ-well counter (Wizard 1480; PerkinElmer, Inc.).
Histology
The mice were deeply anesthetized and perfused transcardially with ice-cold 0.9% saline followed by 4% paraformaldehyde solution. Their brains were removed and afterward were fixed with formalin solution for 6 h, placed in a 30% sucrose solution, and embedded in optimal-cutting-temperature compound (Sakura Finetek Japan Co.). Serial coronal sections (25 μm thick) were obtained with a cryostat and thaw-mounted on glass slides. The rats were perfused with ice-cold 0.9% saline under deep anesthesia and their brains were rapidly frozen in dry ice powder. The brain sections (20 μm thick) were obtained with a cryostat and thaw-mounted on glass slides. For immunostaining, the sections were treated with 2% paraformaldehyde in 0.1 M phosphate-buffered saline for 10 min.
The antibodies used in this study were rabbit IgG (1:500; Cayman Chemical Co.) for COX-1, rabbit IgG (1:500; Cayman Chemical Co.) for COX-2, and rabbit IgG (1:1,000; developed in our laboratory) for the 18-kDa translocator protein.
To characterize cell type, we double-stained the COX-1–positive, COX-2–positive, and translocator protein–positive cells with either the activated microglia/macrophage marker (1:1,000 CD11b, clone OX-42, mouse IgG; AbD Serotec), the astrocyte marker (1:1,000 glial fibrillary acidic protein [GFAP], mouse IgG; Chemicon International, Inc.), or the neuronal marker (1:1,000 neuron-specific nuclear protein, mouse IgG; Chemicon International, Inc.). After overnight incubation of the brain sections with the mixed antibodies at room temperature, the primary antibodies were visualized with either a Cy3-labeled secondary antibody (1:500; Jackson ImmunoResearch) or Alexa Fluor 488 (1:500; Invitrogen). Fluorescent images were captured with a 3-laser confocal microscope (Olympus Optical Co.). The number of immunoreactive cells per section was quantified by counting the number of COX-1–, COX-2–, and OX-42–stained cells and COX-1–/OX-42–costained cell bodies within a 0.4-mm2 area of the striatum.
Statistical Analysis
Statistical analysis was performed using the Student t test. P values less than 0.05 were considered to be statistically significant.
RESULTS
Accumulation of 11C-Ketoprofen Methyl Ester in Brain of COX-1−/− and COX-2−/− Mice
To determine the selectivity of 11C-ketoprofen methyl ester to the COX isoforms, we performed 11C-ketoprofen methyl ester brain imaging on COX-1– and COX-2–deficient mice by ex vivo autoradiography. High accumulations of 11C-ketoprofen methyl ester in the hippocampus, cerebral cortex, and cerebellum were observed in wild-type (COX-1+/+, COX-2+/+) mice, consistent with previous immunohistochemical studies on COX-1 and COX-2 expression (Fig. 1). In all brain regions, accumulation of 11C-ketoprofen methyl ester was significantly lower in COX-1−/− mice than in wild-type (COX-1+/+) mice (Fig. 1A). In contrast, no difference in accumulation was observed between COX-2−/− and wild-type (COX-2+/+) mice (Fig. 1B), indicating that 11C-ketoprofen methyl ester selectively recognizes COX-1 in the brain. Lack of COX-1 and COX-2 proteins in the brains of COX-1−/− and COX-2−/− mice, respectively, was confirmed by immunohistochemistry (Supplemental Fig. 2).
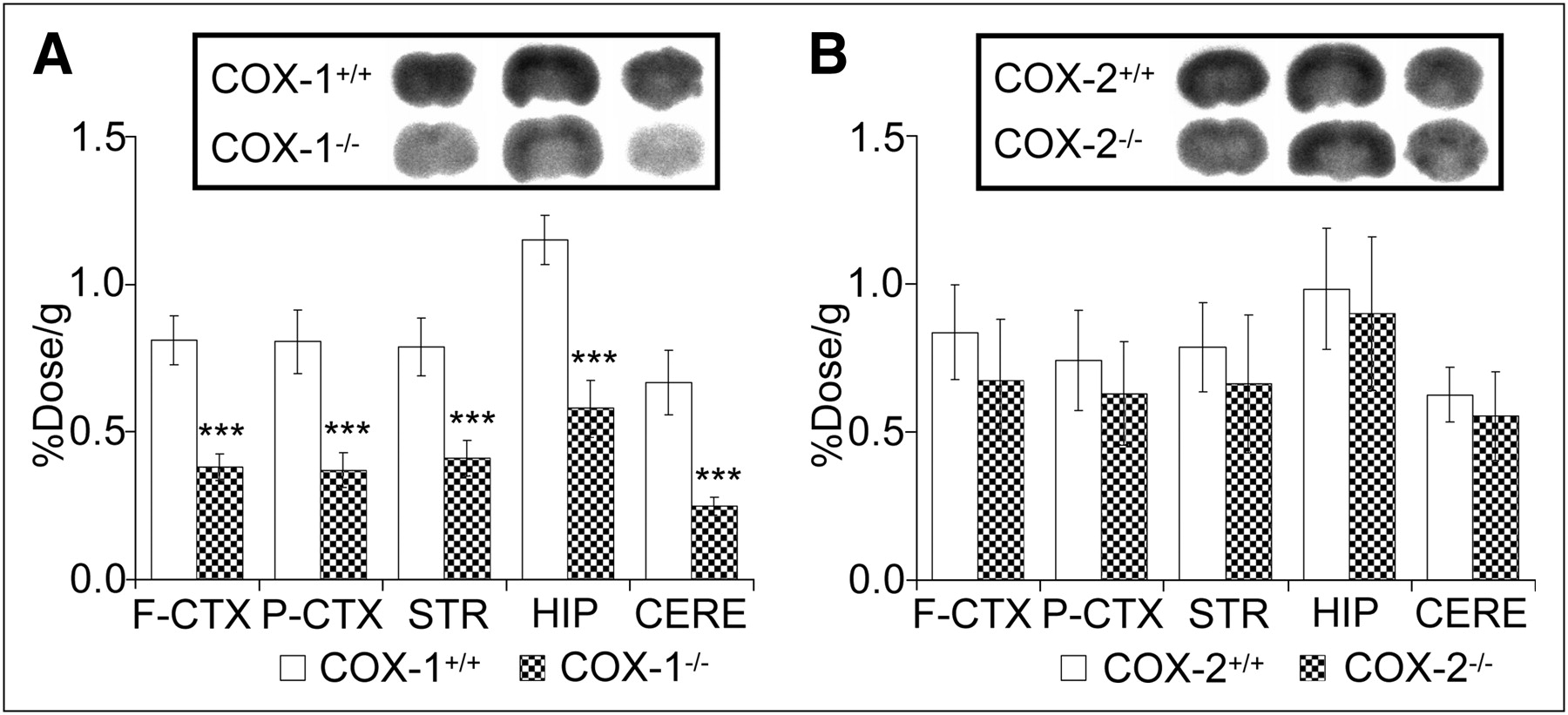
Accumulation of 11C-ketoprofen methyl ester in brain of COX-1−/− and COX-2−/− mice. Ex vivo autoradiography and regional brain tissue uptake of 11C-ketoprofen methyl ester in COX-1+/+ and COX-1−/− mice (A) and COX-2+/+ and COX-2−/− mice (B) was obtained 15 min after injection of 11C-ketoprofen methyl ester. F-CTX = frontal cortex; P-CTX = parietal cortex; STR = striatum; HIP = hippocampus; CERE = cerebellum. Data are mean ± SD (n = 6). ***P < 0.001 vs. wild-type mice.
Time-Dependent Changes of 11C-Ketoprofen Methyl Ester Accumulation in Lipopolysaccharide-Induced Neuroinflammation
Time-dependent changes of 11C-ketoprofen methyl ester after intrastriatal injection of lipopolysaccharide in rats were investigated and were compared with those of 11C-PK11195, a PET probe for activated glial cells. After the lipopolysaccharide injection, a significant increase of 11C-ketoprofen methyl ester accumulation in the ipsilateral striatum was observed at 6 h and at day 1, which is when the accumulation reached its highest level (∼4-fold higher than in the contralateral striatum) (Figs. 2A and 2B). At 7 d after the lipopolysaccharide injection, the accumulation of 11C-ketoprofen methyl ester returned to the basal preinjection level. A slight but significant increase in 11C-ketoprofen methyl ester accumulation was observed only at 6 h in the site of the saline injection (Supplemental Fig. 3), suggesting that needling may drive increased permeability of the blood vessel as an acute inflammatory reaction. On the other hand, a significant accumulation of 11C-PK11195 in the ipsilateral striatum was observed first at day 1, followed by the highest accumulation at day 3 (∼2-fold), and this level lasted until at least day 14 (Figs. 2A and 2C).

Time–radioactivity changes in 11C-ketoprofen methyl ester and 11C-PK11195 accumulation during lipopolysaccharide-induced neuroinflammation. (A) Summed PET image of 11C-ketoprofen methyl ester and 11C-PK11195 from 5 to 45 min in rat brain with hemispheric neuroinflammation induced by intrastriatal lipopolysaccharide (0.5-μg) injection. “Contra” and “LPS” mean contralateral and lipopolysaccharide-injected sides, respectively. (B) Quantification of radioactivity of 11C-ketoprofen methyl ester in contralateral striatum (□) and lipopolysaccharide-injected striatum (▪). (C) Quantification of radioactivity of 11C-PK11195 in contralateral striatum (□) and lipopolysaccharide-injected striatum (▪). Data are SUV mean ± SD (n = 4, 2 of which are obtained from longitudinal PET studies throughout experimental period). KTP-Me = ketoprofen methyl ester. **P < 0.01 vs. contralateral striatum. ***P < 0.001 vs. contralateral striatum.
Identification of Cell Type Associated with COX Expression in Lipopolysaccharide-Induced Neuroinflammation
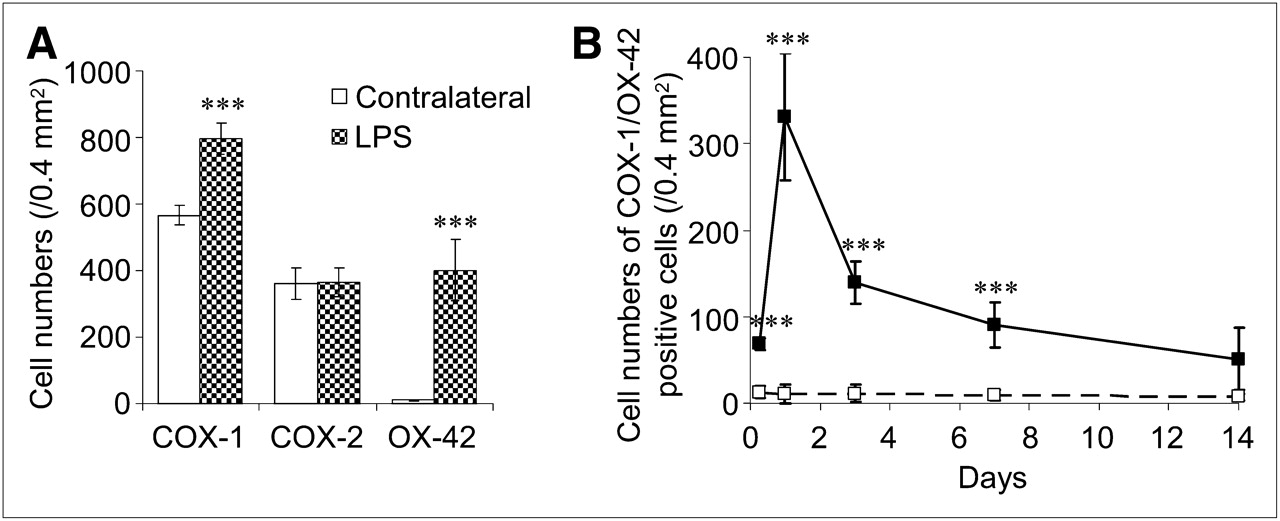
To identify the cell type associated with 11C-ketoprofen methyl ester accumulation in lipopolysaccharide-induced neuroinflammation, we examined double immunostaining of COX-1 and COX-2 with the expression of OX-42, GFAP, and neuron-specific nuclear protein in activated microglia/macrophages, astrocytes, and neurons, respectively. Activation of microglia and macrophages was seen at 6 h and at day 1 after the lipopolysaccharide injection (Fig. 3A). In contrast, though activation of astrocytes was also seen at 6 h after the lipopolysaccharide injection, it lasted to at least 14 d, with a gradual increase in staining intensity. Expression of COX-1 was found in the neurons and activated microglia and macrophages but not in astrocytes (Fig. 3B and Supplemental Fig. 4A). On the other hand, COX-2 expression was observed only in neurons, not in microglia, macrophages, or astrocytes, even after the lipopolysaccharide injection (Fig. 3C and Supplemental Fig. 4B). However, no significant neuronal damage was observed in the core region of the lipopolysaccharide injection site. The population of COX-1–expressing cells was significantly increased, and this increase corresponded to the increased level of activated microglia and macrophages (Fig. 4A). Alternatively, the population of COX-2–expressing cells did not show any changes at any time points examined. The time course of appearance of COX-1–expressing activated microglia and macrophages after the lipopolysaccharide injection closely corresponded to that of 11C-ketoprofen methyl ester accumulation in the injection site of the brain (Fig. 4B), whereas Western blot analysis showed little increase in total protein level of 70-kDa COX-1 in the lipopolysaccharide-injected hemisphere (1.02 ± 0.12-fold from the contralateral side, n = 3) (Supplemental Fig. 5).

Time courses of activation of glial cells and cell-specific expression of COX-1 and COX-2 in lipopolysaccharide-induced neuroinflammation. (A) Representative photomicrographs of OX-42 and GFAP immunohistochemistry in striatum after lipopolysaccharide (0.5-μg) injection. (B) Representative photomicrographs of double immunofluorescent labeling of COX-1 (green) and neuron-specific nuclear protein (a and d; red) and OX-42 (b and e; red) or GFAP (c and f; red) in striatum at day 1 after lipopolysaccharide (0.5-μg) injection. In contralateral striatum (a–c), only neuronal cells with expression of COX-1 are present (arrows in a). In lipopolysaccharide-injected striatum (d–f), COX-1 is expressed in neuronal cells (arrows in d) and in activated microglia and macrophages (arrows in e) but not in astrocytes (f). (C) Representative photomicrographs of double immunofluorescent labeling of COX-2 (green) and neuron-specific nuclear protein (a and d; red), and OX-42 (b and e; red) or GFAP (c and f; red) in striatum at day 1 after lipopolysaccharide (0.5-μg) injection. In contralateral striatum (a–c), only neuronal cells with expression of COX-2 are present (arrows in a). In lipopolysaccharide-injected striatum (d–f), COX-2 is also expressed in neuronal cells (arrows in d) but not in activated microglia and macrophages (e) or astrocytes (f). Scale bars = 50 μm (in A) and 10 μm (in B and C).

Population of immunoreactive cells in striatum after lipopolysaccharide injection. (A) Cells that express COX-1, COX-2, and OX-42 in striatum at day 1 after lipopolysaccharide (0.5-μg) injection. (B) Time-dependent changes in COX-1/OX-42-positive cells in striatum after lipopolysaccharide injection. Numbers of cells in contralateral striatum (□) and in lipopolysaccharide-injected striatum (▪) are shown. Data are expressed as cell numbers (counts/0.4 mm2) and mean ± SD (n = 4). ***P < 0.001 vs. contralateral striatum.
Accumulation of 11C-Ketoprofen Methyl Ester in Quinolinic Acid–Induced Excitotoxic Neurodegeneration
To investigate the possible involvement of COX-1 in the process of excitotoxic neurodegeneration, we performed PET studies with 11C-ketoprofen methyl ester using intrastriatal quinolinic acid–injected rats. Similar to the results of lipopolysaccharide, injecting quinolinic acid also induced a high accumulation of 11C-ketoprofen methyl ester in the ipsilateral striatum at day 1 after injection (Fig. 5). Though the level of 11C-ketoprofen methyl ester accumulation induced by quinolinic acid (SUV, 0.75 ± 0.09) was lower than that observed with lipopolysaccharide (SUV, 0.97 ± 0.16), the ratio of 11C-ketoprofen methyl ester to the contralateral striatum reached approximately 4-fold.

Changes in 11C-ketoprofen methyl ester accumulation in excitotoxic neurodegeneration induced by quinolinic acid. (A) Summed PET image from 5 to 45 min after 11C-ketoprofen methyl ester administration in rat brain 1 d after quinolinic acid (60-nmol) injection. “Contra” and “QUIN” mean contralateral and quinolinic acid–injected sides, respectively. (B) Quantification of radioactivity of 11C-ketoprofen methyl ester in contralateral and quinolinic acid-injected striatum. Data are expressed as SUV and mean ± SD (n = 3). ***P < 0.001 vs. contralateral striatum.
COX Expression in Quinolinic Acid–Induced Neurodegeneration
To identify the expression of COX-1 and COX-2 and their cell type specificity in quinolinic acid–induced excitotoxic neurodegeneration, we performed immunohistochemical analysis of brain samples at day 1 after injection, at which time the number of COX-1–expressed microglia and macrophages peaked to the same level as that of lipopolysaccharide. Quinolinic acid injection produced substantial amounts of neuronal cell loss in the core of the injection site and decreased to approximately 52% in neuron-specific nuclear protein–positive cells, with activation of astrocytes, and microglia and macrophages detected around the injection site. Most of the activated microglia and macrophages showed COX-1 expression, yet the astrocytes showed no COX-1 or COX-2 expression (Supplemental Fig. 6).
DISCUSSION
In this study, we demonstrated that 11C-ketoprofen methyl ester is a suitable PET probe for detecting and monitoring COX-1 expression or activity during neuroinflammation. In addition, by performing PET studies with 11C-ketoprofen methyl ester, we identified the contribution of COX-1 in activated microglia and macrophages during neuroinflammation in vivo.
Previously, PET probes selective for COX-2 rather than COX-1 have been the focus of imaging the inflammatory process, because COX-2 is known to be induced by a variety of inflammatory stimuli, including endotoxins, cytokines, and growth factors (12). However, there has been no successful imaging of inflammation models (13,14). In this study, lipopolysaccharide and quinolinic acid were shown to induce a significantly high accumulation of 11C-ketoprofen methyl ester, which we developed recently, in the injection site of the rat brain. To the best of our knowledge, 11C-ketoprofen methyl ester is the first PET probe targeting COX-1 in neuroinflammation under in vivo conditions.
Ketoprofen, produced by hydrolysis of ketoprofen methyl ester, has a higher affinity to COX-1 than to COX-2, whereby 50% inhibition concentrations in human whole blood cells are 0.047 and 2.9 μM, respectively (25). However, the selectivity of 11C-ketoprofen methyl ester to each COX isoform under in vivo imaging conditions has not yet been clearly determined. The present ex vivo autoradiographic study demonstrated that the accumulation of 11C-ketoprofen methyl ester is significantly low in COX-1−/− mice yet in COX-2−/− does not differ from that in wild-type mice. Therefore, most of the accumulation of 11C-ketoprofen methyl ester in the brain should be derived from its binding to COX-1. In addition to these COX isoforms, a new splice variant of COX-1, referred to as COX-3, which is selectively inhibited by acetaminophen and antipyrine, has been reported (26). Though the selectivity of 11C-ketoprofen methyl ester to COX-3 is not clear, the effect of ketoprofen on methamphetamine-induced hyperthermia in mice has been reported to be different from that of antipyrine (27).
The activation of microglia and macrophages was observed only at the early phase of neuroinflammation, with the largest population observed at day 1 after intrastriatal injection of lipopolysaccharide. Our double immunostaining studies revealed that most of the activated microglia and macrophages (82%) expressed COX-1 but not COX-2. In contrast, astrocytes did not show any expression of either COX-1 or COX-2 throughout the entire neuroinflammation. Though COX-1 is ubiquitously expressed in neuronal cells under normal conditions, any changes in the neuronal cell population and localization of COX-1 in these cells were evident in lipopolysaccharide-induced neuroinflammation. The increased number of COX-1–positive cells in the lipopolysaccharide-injected striatum closely matched that of activated microglia and macrophages. Therefore, we consider that COX-1 may be newly induced with microglial activation or that COX-1–positive macrophages may be infiltrated to the parenchyma during the neuroinflammatory process. Indeed, both proliferation of microglia and invasion of macrophages have been demonstrated in the brain during neuroinflammation induced by the intraperitoneal or intrastriatal injection of lipopolysaccharide in mice (28,29).
We identified the expression of COX-2 only in neuronal cells, and induction of COX-2 expression was not observed in any brain cells at any time points after lipopolysaccharide injection, although it has been reportedly induced in inflammatory stimuli including lipopolysaccharide, mostly with cultured cells in vitro (30). Intracranial injection of lipopolysaccharide into the substantial nigra is reported to cause dopaminergic neuronal death with a transient increase in COX-2 expression (31), but the cell types expressing COX-2 have not yet been identified. The authors of that study used an approximately 4-fold higher dose of lipopolysaccharide than we used, and so the dosage or the site of injection may contribute to COX-2 induction in neural cells under in vivo conditions. In agreement with our results, COX-2 expression in neuronal cells but not in microglia or astrocytes was also reported in animal models and in patients with neurodegeneration such as that caused by Parkinson disease and Alzheimer disease (32,11).
11C-ketoprofen methyl ester showed high accumulation into the inflamed area at the early phase after lipopolysaccharide injection, and the time dependency corresponded well with the population of COX-1–expressing activated microglia and macrophages. The increased number of COX-1–expressing microglia and macrophages was approximately 40% of the total number of COX-1–expressing cells, which is not significant in protein level, though increased 11C-ketoprofen methyl ester accumulation was much higher than that of the corresponding radioactivity related to this increase (3.7-fold from the basal level). Thus, not only expression but also elevated enzymatic activity of COX-1 might be related to the changes in radioactivity of 11C-ketoprofen methyl ester observed in the PET study.
Recently, it has been reported that pharmacologic inhibition and gene disruption of COX-1 in mice decreases the activation of microglia and astrocytes induced by the intracerebroventricular injection of lipopolysaccharide with reduction of proinflammatory cytokines, chemokines, and oxidative stress (33). However, COX-2–deficient mice show exacerbation of neuronal damage with an increase of proinflammatory agents and oxidative stress in response to lipopolysaccharide (34). In addition, neuroinflammation-induced blood–brain barrier breakdown induced by lipopolysaccharide has also been shown to be attenuated by COX-1 inhibition yet was not prevented by COX-2 inhibition (35). Supporting our data, these studies indicate that COX-1 plays proinflammatory roles in neuroinflammatory processes. It has also been reported that COX-1–positive microglia are largely present in neuronal diseases, such as Alzheimer disease, Parkinson disease, and traumatic brain injury, indicating the involvement of COX-1 via activation of microglia in the pathologic process of these neurodegenerative disorders (8–11). Therefore, COX-1 might be a critical target for the diagnosis and therapeutic treatment of neurodegenerative disorders.
The elevation of 11C-ketoprofen methyl ester accumulation and COX-1–expressing activated microglia and macrophages were observed in neuroinflammation induced by both lipopolysaccharide and quinolinic acid, though the mechanisms of these compounds were quite different. It is well established that lipopolysaccharide is a stimulant of the innate immune system, recognized by TLR4, and induces a robust inflammatory response by activating phagocytic cells such as microglia (16,17). Injection of quinolinic acid induced significant neuronal death in the rat striatum, along with activation of microglia and macrophages expressing COX-1—a result that is similar to the results obtained for lipopolysaccharide. However, quinolinic acid has activated astrocytes different from lipopolysaccharide in the early phase, and no expression of COX-1 or COX-2 in these astrocytes was observed. Quinolinic acid is reported to induce direct damage of neuronal cells and astrocytes by its excitatory effect, with microglial activation seemingly a secondary reaction of this cell death (18,19). The involvement of COX-2 rather than COX-1 in neuronal damage induced by quinolinic acid has previously been reported (36). Inhibition of COX-2 but not COX-1 was found to provide structural and functional protection against quinolinic acid–induced neurodegeneration. Different from our experimental conditions, an almost 4-fold higher dose (225 nmol) of quinolinic acid was used in that study, and the authors evaluated the effects at 27 d after the quinolinic acid injection. Therefore, COX-2 might be involved in the later phase of inflammation associated with reactive gliosis, caused by a robust insult, rather than the early phase of initiation of proinflammatory factors.
The time-dependent changes of 11C-ketoprofen methyl ester and 11C-PK11195 were quite different in the rat striatum after the injection of lipopolysaccharide. High accumulation of 11C-ketoprofen methyl ester was observed in the early phase, corresponding well to the time course of activation of microglia and macrophages, as demonstrated by immunohistochemistry. Different from our results, the results of some animal model studies of ischemia and also lipopolysaccharide injection with a higher dosage (100-fold) showed strong and prolonged activation of microglia after injury (37,38). The time course of activation of microglia and macrophages might depend on the nature or intensity of the injury. In contrast, the high accumulation of 11C-PK11195 started slowly and lasted until at least 14 d afterward. Because 11C-PK11195 is known to be specific to translocator protein, which is highly expressed in activated microglia and astrocytes (39,40), the time course of 11C-PK11195 might correspond to changes in both activation of microglia/macrophages and astrocytes. As a preliminary result, we also observed the appearance of translocator protein/OX-42– and translocator protein/GFAP–positive cells at the early and late phases, respectively (Supplemental Fig. 7). Therefore, 11C-ketoprofen methyl ester is highly responsible for and sensitive to the neuroinflammatory processes targeting COX-1 in activated microglia and macrophages.
Hydrolysis of 11C-ketoprofen methyl ester possibly influences its brain accumulation. However, in rats, 11C-ketoprofen methyl ester that incorporates into the brain is hydrolyzed rapidly (15), and levels of 11C-ketoprofen in normal and inflamed brain tissues at 2 min after 11C-ketoprofen methyl ester administration have shown no significant difference (70.3% ± 1.4% vs. 80.5% ± 6.5%, n = 3, P > 0.05) at day 1 after lipopolysaccharide injection. Although further studies on quantitative analysis with kinetic models are required for clinical application, 11C-ketoprofen methyl ester could act as a valuable tool for investigating the role of COX-1 and for diagnosing the pathologic process of neurodegenerative disorders.
CONCLUSION
We have identified 11C-ketoprofen methyl ester as a COX-1–selective PET probe and have succeeded in visualizing the expression of COX-1 in microglia and macrophages during the neuroinflammatory process in the living brain. Time-dependent in vivo expression of COX-1 in activated microglia and macrophages during neuroinflammation, which was revealed by our PET study of 11C-ketoprofen methyl ester in rats, strongly indicates a specific action of COX-1 in the neuroinflammatory process. COX-1 may play a crucial role in the pathology of neuroinflammation and might be a critical target for the diagnosis and therapy of neurodegenerative disorders.
DISCLOSURE STATEMENT
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported in part by a consignment expense for the Molecular Imaging Research Programs titled “Research Base for Exploring New Drugs” and “Kobe Cluster” from the Ministry of Education, Culture, Sports, Science and Technology of the Japanese government; a Health and Labour Sciences Research Grant for Research on Regenerative Medicine for Clinical Application; and a Grant-in-Aid for Young Scientists (B) of the Japan Society for the Promotion of Science. No other potential conflict of interest relevant to this article was reported. We thank Masahiro Kurahashi (Sumitomo Heavy Industry Accelerator Service Ltd.), Yumiko Katayama, Miki Goto, Yasuhiro Wada, and Emi Hayashinaka (RIKEN) for their assistance with the PET study.
- © 2011 by Society of Nuclear Medicine
REFERENCES
- Received for publication October 13, 2010.
- Accepted for publication February 28, 2011.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Chemogenetic activation of target neurons expressing insect Ionotropic Receptors in the mammalian central nervous system by systemic administration of ligand precursors
- Synthesis and Preclinical Evaluation of 18F-Labeled Ketoprofen Methyl Esters for Cyclooxygenase-1 Imaging in Neuroinflammation
- Enhanced aversive memory retrieval by chemogenetic activation of locus coeruleus norepinephrine neurons
- Evaluation of Two Potent and Selective PET Radioligands to Image COX-1 and COX-2 in Rhesus Monkeys
- Detection of Cyclooxygenase-1 in Activated Microglia During Amyloid Plaque Progression: PET Studies in Alzheimers Disease Model Mice
- Neuroinflammation in Patients with Chronic Fatigue Syndrome/Myalgic Encephalomyelitis: An 11C-(R)-PK11195 PET Study