


Abstract
Mammalian target of rapamycin (mTOR) is an evolutionally conserved serine/threonine kinase that integrates signals from multiple pathways, including nutrients (e.g., amino acids and glucose), growth factors (e.g., insulin and insulinlike growth factor 1), hormones (e.g., leptin), and stresses (e.g., starvation, hypoxia, and DNA damage) to regulate a wide variety of eukaryotic cellular functions, such as translation, transcription, protein turnover, cell growth, differentiation, cell survival, metabolism, energy balance, and stress response. Dysregulation of the mTOR pathway is closely associated with cancers and other human diseases. Thus, mTOR is of considerable interest in view of its potential as a therapeutic drug target. However, only limited success has been achieved in clinical applications of mTOR inhibitors because of the inherent complexity in the regulation and function of mTOR. Emerging new developments in this area, such as novel readouts (potential biomarkers) for mTOR activity, dynamic assembly and translocation of the mTOR complex, cross-regulation between mTOR complex 1 and mTOR complex 2 via inter- and intracomplex loops, new mTOR regulators, and new inhibitors, are providing insights that may help overcome these challenges. The introduction of innovative imaging strategies is also expected to give rise to breakthroughs in understanding mTOR network complexity and mTOR inhibitor action by visualizing the regulation and function of mTOR.
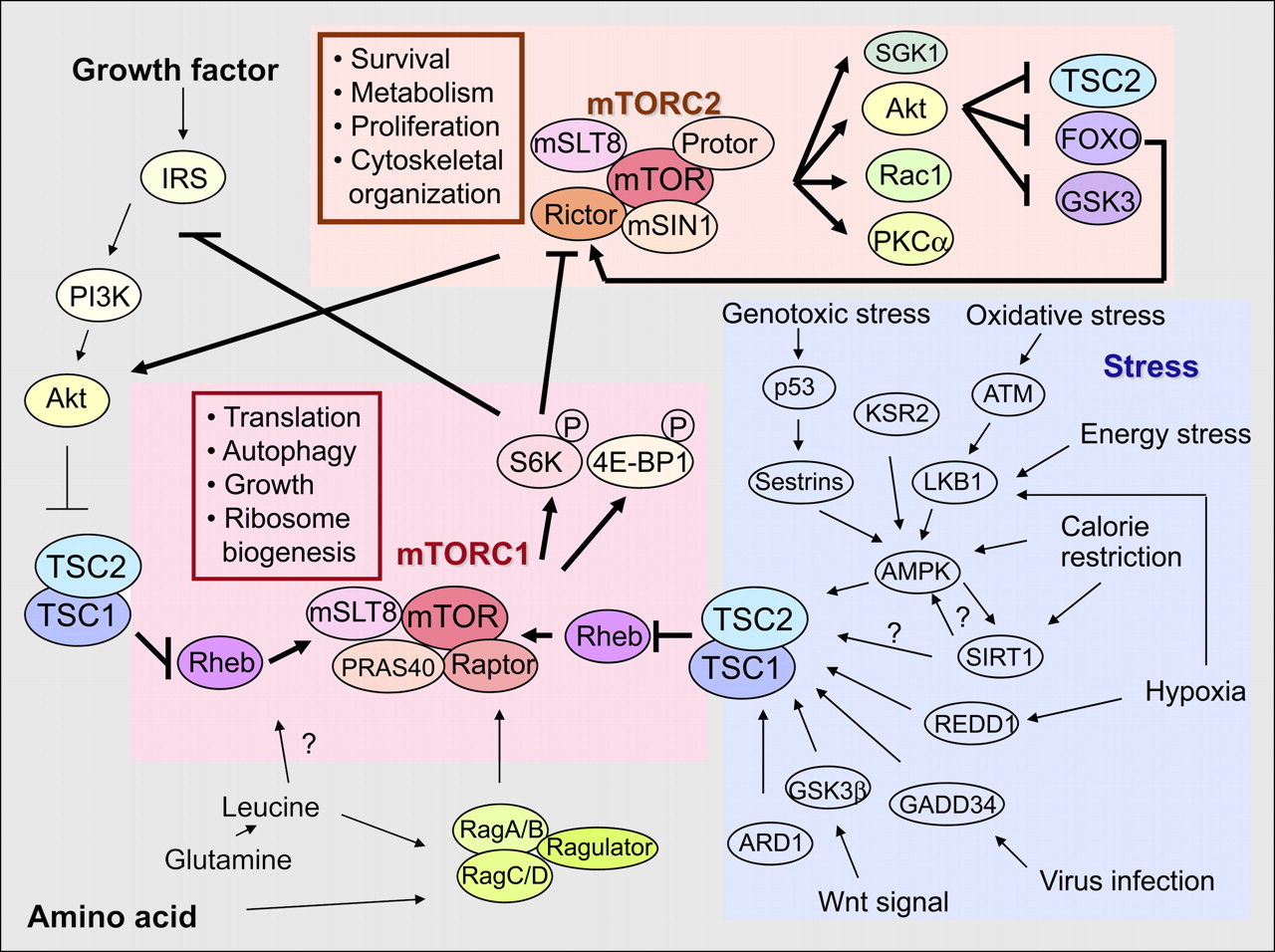
Mammalian target of rapamycin (mTOR) is one of the protein kinases related to phosphatidylinositol 3-kinase and structurally contains an N-terminal HEAT repeat domain and a C-terminal kinase domain flanked by FAT and FATC domains (Fig. 1). mTOR exists in 2 functionally and structurally distinct complexes, rapamycin-sensitive mTOR complex 1 (mTORC1) and rapamycin-insensitive mTOR complex 2 (mTORC2), which becomes rapamycin-sensitive after prolonged rapamycin treatment (Fig. 2). mTORC1 phosphorylates S6K and 4E-BP1 (which are used as a readout for mTORC1 activity) and regulates translation, autophagy, growth, lipid biosynthesis, mitochondria biogenesis, and ribosome biogenesis, whereas mTORC2 phosphorylates SGK1, Akt (S473 phosphorylation is used as a readout for mTORC2 activity), Rac1, and PKCα and regulates survival, metabolism, proliferation, and cytoskeletal organization (1,2).
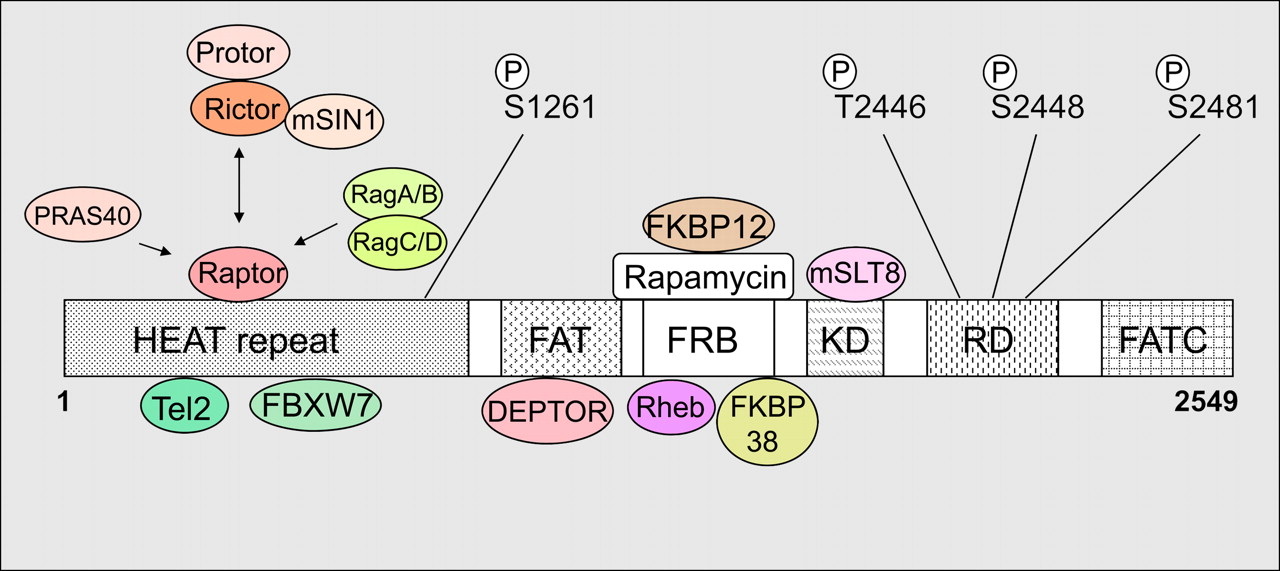
mTOR, its interacting proteins, and phosphorylation sites. DEPTOR = DEP domains and its specific interaction with mTOR; FAT = FRAP, ATM, TRRAP; FATC = FRAP ATM TRRAP carboxy terminus; FKBP = FK506 binding protein; FKBP12 = 12-kDa immunophilin FK506-binding protein; FRB = FKBP12 rapamycin-binding; FXBW7 = F-box and WD repeat domain–containing 7 (also known as hCDC4, FBW7, and hAGO); HEAT = Huntington, elongation factor 3, PR65/A, TOR; KD = kinase domain; mSLT8 = mammalian homolog of lethal with sec-13 gene 8; PRAS40 = proline-rich Akt substrate, 40 kDa; Protor = protein observed with Rictor-1; Rag = ras-related GTP-binding protein; Raptor = regulatory associated protein of mTOR; RD = regulatory domain; Rheb = Ras homolog enriched in brain; Rictor = rapamycin-insensitive companion of mTOR. Four mTOR phosphorylation (P) sites (S1261, T2446, S2448, and S2481) and mTOR-interacting proteins are indicated.

mTOR complexes and mTOR signaling network. ARD1 = arrest-defective protein 1; ATM = ataxia telangiectasia, mutated; 4E-BP1 = eukaryotic initiation factor 4E-binding protein 1; FOXO = forkhead box O; GADD34 = growth arrest and DNA damage; GSK3 = glycogen-synthase-kinase-3; IRS = insulin receptor substrate; KSR2 = kinase suppressor of Ras 2. PI3K = phosphoinositide-3-kinase; PKC = protein kinase C; Rac1 = ras-related C3 botulinum toxin substrate 1; REDD1 = regulated in development and DNA damage response genes 1; SGK1 = glucocorticoid inducible kinase-1; S6K = ribosomal protein S6 kinase; TSC1/2 = tuberous sclerosis 1/2. Selected components and functions of both mTOR1 and mTORC2 are indicated, and both mTORC1 and mTORC2 additionally interact with DEPTOR, which usually inhibits the activity of both complexes. Growth factor such as insulin stimulates mTORC1 (and probably also mTORC2), leading to Akt activation to inhibit TSC2, a GTPase activating protein for Rheb. Amino acid also activates mTORC1 through glutamine-leucine and Rag–Ragulator complex, which is required for full activation of mTORC1 by growth factor. Little is known about activation mechanism of mTORC2. Feedback loop by S6K-IRS or S6K-Rictor exists in mTOR signaling. In contrast, cellular stress activates TSC2 and inhibits mTOR pathway.
mTOR interacts with Tel2 (also known as TELO2 and hCLK2) and FXBW7 (Fig. 1). Deletion of Tel2 results in destabilization of mTOR, whereas FBXW7 targets mTOR for ubiquitin-proteasome–dependent degradation (3,4). mTOR also interacts with Raptor, which is a specific component of mTORC1, through binding to the N-terminal HEAT domain (Figs. 1 and 2). Raptor functions as a scaffold protein to recruit S6K and 4E-BP1 to promote protein synthesis through the direct phosphorylation of S6K and 4E-BP1 by mTORC1 (Fig. 2) (5). Overexpression of Rictor, a specific component of mTORC2, disrupts mTOR–Raptor interaction. This indicates that Rictor may compete with Raptor in binding to mTOR through the HEAT domain. DEPTOR, an inhibitory protein for mTOR, binds through the FAT domain of mTOR, and overexpression of DEPTOR results in the suppression of S6K by inhibiting Akt, whereas loss of DEPTOR activates S6K and Akt.
Rapamycin, the best-characterized mTOR inhibitor, binds to FKBP12, which in turn targets mTOR through its FRB domain, resulting in an inhibition of mTOR function. Mutation of a conserved serine (aa 2035 in human mTOR) in the FRB domain confers resistance to rapamycin (6). Amino acid regulates dynamic assembly and translocation of mTORC1 with FKBP38 and Rag guanosine triphosphatase (GTPase) (5). Mitochondrial membrane protein FKBP38 is an endogenous inhibitor of mTOR and binds to the FRB domain of mTOR (7). mTOR–FKBP38 interaction is increased by amino acid or serum starvation, leading to inhibition of mTOR activity. The Rheb GTPase protein also binds to the FRB domain of mTOR and is involved in mTOR activation by amino acids. On amino acid stimulation, 4 members of the Rag subfamily of Ras small GTPases—RagA, RagB, RagC, and RagD—bind to Raptor directly, and Rag-bound mTORC1 translocates to Rab7 (a GTPase required for transporter degradation)-positive perinuclear vesicular structures, where Rheb localizes. This relocalization enables mTORC1 binding to Rheb, leading to mTORC1 kinase activation. Ragulator recruits Rag family proteins and mTORC1 to lysosomes to activate mTORC1 (8). mLST8 binds to the kinase domain of mTOR and strongly enhances mTOR signaling. Knockout studies demonstrated that mSLT8 is required to maintain the Rictor-mTOR, but not the Raptor-mTOR, interaction regardless of the fact that both mTORC1 and mTORC2 contain mSLT8, indicating the mTORC2-specific role of mSLT8 (9).
PHOSPHORYLATION OF MTOR
There are 4 characterized phosphorylation sites in mTOR (Fig. 1). T2446 is regulated by nutrient availability and is probably phosphorylated by adenosine monophosphate kinase (AMPK). S2448 is phosphorylated by S6K, which directly reflects amino acid and nutrient status. S2481 was known as a rapamycin-insensitive autophosphorylation site. mTORC1 contains mainly mTOR phosphorylated at S2448, whereas mTORC2 contains predominantly mTOR phosphorylated at S2481, sensitive to short versus prolonged rapamycin treatment, respectively (10). The finding that mTORC1 also contains mTOR phosphorylated on S2481 in the same cell line and that the phosphorylation on S2481 in both mTORC1 and mTORC2 is sensitive to wortmannin, a phosphatidylinositol 3–kinase inhibitor (11), indicates that insulin signals via phosphatidylinositol 3–kinase to promote mTORC1 and mTORC2-associated mTOR autophosphorylation on S2481. These discrepancies between previous and more recent work are likely due to the fact that existence of mTORC1 and mTORC2 with different sensitivities to rapamycin was apparent only later. S1261 within the HEAT domain is phosphorylated by insulin/phosphatidylinositol 3–kinase in an amino acid–dependent, rapamycin-insensitive, and autophosphorylation-independent manner. S1261 phosphorylation promotes the phosphorylation of S6K and 4E-BP1, and mutation on S1261 attenuates phosphorylation on S2481.
FEEDBACK LOOPS
Feedback loops are an important mechanism to regulate mTOR signaling (Fig. 2). mTORC1-stimulated S6K inhibits IRS through multiple phosphorylation sites on IRS, resulting in its degradation. S6K also phosphorylates Rictor at T1135 (12). Mutation at T1135 on Rictor results in increased phosphorylation of Akt at S473 and its downstream target, FOXO1/3a and GSK3α/β. Knockdown of Rictor results in an unchanged or a slight increase in the phosphorylation of S6K, indicating that mTORC1 and mTORC2 cross-regulate their activities through S6K and Rictor. Stress-activated FOXO increases the amount of Rictor and upregulates mTORC2 activity but downregulates mTORC1 activity (13). In this regard, identification and characterization of mTORC1 and mTORC2 were an important step toward understanding and visualizing the behavior of the mTOR complex structurally and functionally.
INHIBITORY MECHANISMS OF MTOR SIGNALING PATHWAY
Under low energy (high adenosine monophosphate–to–adenosine triphosphate ratio), AMPK is activated in an LKB1-dependent manner to phosphorylate TSC2 (Fig. 2) (2). AMPK also directly phosphorylates Raptor at S792 in an LKB1-dependent manner, which is required for the inhibition of mTORC1 and growth arrest under energy stress. Under hypoxic conditions, hypoxia-inducible factor 1–mediated upregulation of REDD1 and REDD2 proteins by hypoxia leads to the activation of TSC1/2 in an LKB1-AMPK–independent manner. TSC2 has been reported to be a substrate of GSK3, and activation of the Wnt pathway stimulates the mTOR pathway by phosphorylating TSC2 by GSK3.
Under high levels of cellular reactive oxygen species, ATM, a regulator of the DNA damage response, activates TSC2 via the LKB1-AMPK pathway to inhibit the mTOR pathway and to induce autophagy (14). Rapamycin inhibits elevated reactive oxygen species and mTOR activity in ATM−/− cells, indicating cross-talk between the DNA damage response and energy metabolic pathway.
GADD34 is induced by almost all cellular stresses and binds and dephosphorylates TSC2. GADD34−/− cells are more sensitive to glucose starvation and virus infection than WT cells, resulting in apoptosis due to inability to suppress the mTOR pathway (15). Treatment of rapamycin indeed suppresses apoptosis in GADD34−/− cells, suggesting that stress stimuli inhibit the mTOR pathway through the GADD34-TSC2 axis.
Recently, an acetylation-mediated TSC2 regulation was reported (16). ARD1, an acetyltransferase and a putative tumor suppressor, binds, acetylates, and stabilizes TSC2, leading to inhibition of the mTOR pathway. The expression of ARD1 correlates with that of TSC2 in multiple tumor types, and loss of heterozygosity at the ARD1 locus was observed in human breast, lung, pancreatic, and ovarian cancer samples.
KSR2, a regulator of extracellular signal-regulated kinase 1/2 (a mitogen-activated protein kinase), binds and modulates the activity of AMPK. KSR2 regulates AMPK-dependent glucose uptake and fatty acid oxidation, and KSR2 knockout mice show decreased fatty acid oxidation and thermogenesis resulting in obesity (17).
Three sestrin family proteins inhibit the mTOR pathway through the AMPK-TSC2 axis. p53 target genes Sestrin1 and Sestrin2 are induced on oxidative stress and DNA damage and bind and activate AMPK, resulting in TSC2-dependent inhibition of the mTOR pathway (18). The expression of mammalian Sestrin3 is regulated by Akt in a FOXO3a-dependent manner and activates AMPK and regulates cellular reactive oxygen species accumulation (13,19). Increased reactive oxygen species caused by accelerated oxygen consumption in Akt1/2 knockout cells was reduced by knockdown of Sestrin3, indicating that Sesn3 plays an important role in the regulation of cellular reactive oxygen species mediated by Akt and FOXO (19).
Sirtuin 1 (SIRT1) is a nicotinamide adenine dinucleotide (NAD+)–dependent deacetylase that has been implicated in regulation of the mTOR pathway. Although SIRT1 deacetylates LKB1 for the LKB1-AMPK activation, AMPK may regulate SIRT1 activity by increasing intracellular NAD+. AMPK kinase activity is required to trigger SIRT1-dependent response to exercise and fasting, but it remains unknown whether AMPK is required for fasting-induced activation of SIRT1 and deacetylation of its targets (20). Further studies will be required to determine the interdependence of AMPK and SIRT1 on the mTOR pathway.
MTOR INHIBITORS IN CLINICAL TRIALS AND THE NEED FOR COMPANION BIOMARKERS
Because more than 80% of human cancers acquire hyperactivation of the mTOR pathway, rapamycin has been expected to have powerful anticancer effects. The effects of the combined use of rapalogs with other anticancer agents or rapalogs alone are under investigation in several human cancers, such as brain, breast, and other solid tumors (21,22). More complex than expected, so far rapalogs have achieved only limited success in cancer treatment, perhaps because of cell type–dependent sensitivity to rapamycin, feedback loops, rapamycin-independent mTOR function (e.g., phosphorylation of 4E-BP1), development of resistance to rapamycin, inadequate drug delivery to tumor targets, and insufficient understanding of the molecular mechanism by which rapamycin inhibits mTOR and cell growth (even today, exactly how rapamycin perturbs mTOR function is not completely understood). Dual mTOR–phosphatidylinositol 3–kinase inhibitors, such as NVP-BEZ235 and PI-103, and a different class of mTOR inhibitors (e.g., Torin1, PP242, PP30, Ku-0063794, WAY-600, WYE-687, WYE-354, and CC-223) that act through the canonic kinase inhibitor mechanism by targeting the adenosine triphosphate–binding pocket of the mTOR kinase domain, have been developed, and their anticancer effects are being investigated (23).
When one is considering effective therapy with rapalogs and other anticancer agents, the concept of synthetic lethality may help overcome many of the problems described above (24,25). We envision that these agents may be used in combination therapy with rapamycin to yield the following benefits: to sensitize a tumor's response to rapamycin, to provide a synthetic lethal strategy against rapamycin-insensitive tumors, and to prevent or delay the development of rapamycin resistance in tumors (analogous to the successful use of drug cocktails for treating HIV infection).
Several biomarkers have been developed to monitor the effects of mTOR inhibitors. These include measurements by Western blot or immunohistochemistry of S6K and 4E-BP1 phosphorylation and of various immunocytokines, including interleukin 2, 4, and 10. Because these methods may lack the required selectivity and sensitivity, there is a clear need for the identification and validation of new biomarker sets to predict and monitor responses to mTOR inhibitors. The combined use of different classes of biomarkers may be needed to accurately predict responses to mTOR inhibitors (26). In particular, similar to the revolutionary use of 18F-FDG (a glucose analog) (27), and 3′-deoxy-3′-18F-fluorothymidine (18F-FLT) (a thymidine analog) (28), discovery of endogenous small-molecule surrogate biomarkers should enable the development of real-time noninvasive molecular imaging agents that will find immediate utility in the clinic both for drug therapy monitoring and for new drug development and trials. PET with 18F-FDG and 18F-FLT for monitoring tumor responses to mTOR inhibitors has been evaluated in preclinical and clinical studies. It has been shown in mice that rapamycin significantly reduced the 18F-FDG and 18F-FLT uptake in human U87 glioma xenografts (29). In another study, the mTOR inhibitor everolimus (RAD001) induced a strong inhibition of 18F-FLT uptake in human SKOV3 ovarian cancer xenografts (30). Recently, promising results were observed in a phase I clinical trial with the mTOR inhibitor RAD001 in glioma patients. In this study, 18F-FDG PET revealed partial metabolic responses in a subset of patients. Collectively, these preliminary results are encouraging, and they should provide the impetus for more extensive studies to validate such PET biomarkers for monitoring therapeutic interventions using mTOR inhibitors.
Acknowledgments
We thank Caius Radu for critical reading and comments on the manuscript, and we thank the American Cancer Society (RSG-07-035-01-CCG) and the National Institutes of Health (R01 CA124974 and R21 CA149774) for funding support.
- © 2011 by Society of Nuclear Medicine
REFERENCES
- Received for publication July 1, 2010.
- Accepted for publication February 23, 2011.





{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- TTBK2 controls cilium stability through actin and the centrosomal compartment
- mTOR inhibition impairs extinction memory reconsolidation
- Akt Pathway Inhibition of the Solenopsin Analog, 2-Dodecylsulfanyl-1,-4,-5,-6-tetrahydropyrimidine
- Nrf2 Transcription Factor Can Directly Regulate mTOR: LINKING CYTOPROTECTIVE GENE EXPRESSION TO A MAJOR METABOLIC REGULATOR THAT GENERATES REDOX ACTIVITY
- Delineating the mTOR Kinase Pathway Using a Dual TORC1/2 Inhibitor, AZD8055, in Multiple Myeloma
- Hitting the target: where do molecularly targeted therapies fit in the treatment scheduling of neuroendocrine tumours?