


Abstract
Dementia is a highly prevalent problem causing considerable disability and mortality and exacting great costs to individuals, their families, and society. The 4 most common neurodegenerative disorders that cause dementia—Alzheimer disease, frontotemporal dementia, dementia with Lewy bodies, and dementia in Parkinson disease—have different underlying etiologies and pathogenetic mechanisms. There is a great need for early diagnostic markers; functional brain imaging may therefore assist in the detection and differential diagnosis of dementia due to neurodegenerative diseases. Functional imaging such as PET allows in vivo imaging of functional brain activity indicating cerebral blood flow and cerebral glucose metabolism, and PET allows imaging of neurotransmitter activity, including that of the cholinergic, dopaminergic, and serotonergic systems. New PET neuroimaging tracers are being developed for detecting pathologic parameters such as amyloid plaque and microglial activity. The development of molecular imaging is important for early diagnosis of dementia, selection of patients for therapies, and evaluation of therapies.
Neurodegenerative diseases are a large group of neurologic disorders. By definition, a neurodegenerative disease is one with selective and progressive loss of specific populations of neurons for reasons that in most cases remain unknown. The most common neurodegenerative disorders are associated with intracellular or extracellular deposits of abnormal proteins. The particular population of neurons that are vulnerable in each disorder determines the clinical presentation, and each disorder is defined by a specific combination of clinical, pathologic, and biochemical features. In the present review, we discuss the application of neuroimaging techniques such as PET in neurodegenerative disorders causing dementia, including Alzheimer disease (AD), frontotemporal dementia (FTD), dementia with Lewy bodies (DLB), and dementia in Parkinson disease (PDD).
AD is presently considered the most prevalent neurodegenerative disorder and is characterized by symptoms such as progressive loss of memory, especially episodic memory, as well as loss of other cognitive functions. Data suggest that the number of AD patients worldwide will increase from 26.6 million in 2006 to 106.8 million in 2050 (1). To tackle both the increasing social costs and the burden on patients and their families, great efforts are being made to develop early diagnostic markers of AD. These will enable early drug intervention and, it is hoped, lead to a cure for AD. The neuropathologic hallmarks of AD, including β-amyloid (Aβ) deposition in extracellular plaques and vascular walls, the accumulation of intracellular neurofibrillary tangles (NFTs), synaptic reduction, neuronal loss, and volume loss (atrophy), probably occur many years before cognitive symptoms (2). Aβ is considered a primary cause of AD, but there is a whole cascade of other ongoing processes, including oxidative stress, inflammatory reactions, microglial activation, tau phosphorylation, and neurotransmitter impairment, leading to cognitive impairment (3). Some research criteria for the diagnosis of AD were proposed by Dubois et al. in 2007 (4). These criteria suggest the presence of early episodic memory impairment with at least one abnormal biomarker detectable by structural neuroimaging with MRI, molecular neuroimaging with PET, or cerebrospinal fluid (CSF) analysis of Aβ or τ-proteins. Further studies are needed to validate the clinical significance of these suggested diagnostic criteria.
FTD is a clinically, genetically, and neuropathologically heterogeneous group of diseases. The neurodegenerative process in FTD may be focal over several years, affecting either the left or the right hemisphere and involving predominantly frontal and temporal areas. The clinical symptoms of FTD are often subtle and include personality changes. Cognitive disturbances typical of FTD include attention deficits, impaired executive function, and language disturbances. Several types of neuropathologic changes underlie the clinical syndrome of FTD; all patients have in common the lobar atrophy, neuronal loss, and gliosis. Other neuropathologic processes include the presence or absence of τ-protein, ubiquitin, and the predominant τ-isoform, as detected by immunohistochemistry (5). Recent studies indicate that most FTD patients show brain pathology with ubiquitin-positive inclusions whereas some FTD patients show τ-positive inclusions (6). Familial FTD is common, and genetic mutations have been demonstrated on chromosome 17 in the τ or progranulin genes (6).
The core features for the diagnosis of DLB are fluctuating cognition with pronounced variations in attention and alertness, recurrent visual hallucinations that are typically well formed and detailed, and spontaneous features of parkinsonism (7), as well as visual spatial problems. The pathologic diagnosis of DLB requires demonstration of cortical Lewy bodies, and its staging is determined by the density and distribution of these inclusions, though in reality this staging correlates only weakly with the severity or duration of dementia during life (8). In addition to Lewy body pathology, a significant number of patients with DLB have shown concomitant AD pathology (amyloid plaques and neurofibrillary tangles) and significant amyloid deposition in the striatum at postmortem examination (9).
Many patients with PD also develop dementia. The reported prevalence of dementia in PD varies from 4% to 93% and averages around 40% (10). The dementia associated with PD is characterized by impairment of short-term recall, attention, visuospatial functions, and executive functions such as decision making. In addition to manifesting cognitive impairment, PD patients can manifest personality and behavioral changes along with fluctuating confusion and visual hallucinosis. Although most clinical features of PDD overlap with DLB (11), recent guidelines defined the 1-y rule for distinction between them according to the onset of cognitive dysfunction in relation to extrapyramidal motor symptoms (7,12). Pathologically, PD is characterized by the presence of cortical Lewy bodies and degeneration of subcortical nuclei including the dopaminergic medial substantia nigra, the cholinergic nucleus basalis of Meynert, the noradrenergic locus ceruleus, and serotonergic median raphe, along with other brain stem nuclei. The presence of Aβ has occasionally been reported in idiopathic PD, though this is more commonly a feature in PD patients who have developed later dementia (PDD) (13).
Mild cognitive impairment (MCI) is considered a transitional stage between normal aging and dementia disorder, especially in AD. MCI is a heterogeneous syndrome characterized by memory impairment or impairment in another cognitive domain but with preserved activities of daily living (14). Amnestic MCI (∼30% of MCI patients) refers to the subtype that involves impairment primarily of the memory component, either alone (single-domain) or in conjunction with impairment of other cognitive domains (multiple-domain). Clinical cohorts suggest that about 12% of patients with the amnestic form of MCI progress to AD each year, and up to 80% have progressed to AD after 6 y (15). However, some MCI patients are likely to develop other form of dementia such as FTD or DLB (16).
Pathologic studies have shown that it is possible to find mixed dementia (i.e., AD plus vascular lesions) in elderly people (17). Studies also have suggested that AD and vascular lesions may interact with each other to increase the probability of dementia (17,18). However, the clinical features typical of AD (e.g., progressive cognitive decline) may also be found in vascular dementia.
Quantitative imaging of functional and pathologic processes in the living human brain has become feasible through the development of PET techniques. PET is a noninvasive tomographic method for imaging the regional distribution of radioactive tracers. PET may facilitate early diagnosis, differential diagnosis, and evaluation of drug treatment in patients with dementia. PET tracers that have been developed for studies of functional activity, neurotransmitter activity, and pathologic processes in different dementia disorders (Tables 1 and 2) are reviewed in this paper.
PET Ligands Used to Measure Functional Activity and Neurotransmitters in Patients with Different Dementias
PET Ligands Used to Measure Pathologic Markers in Patients with Dementia
BRAIN GLUCOSE METABOLISM
The cerebral metabolic rate of glucose (CMRglc) in humans is studied by means of 18F-FDG and may reflect synaptic activity in different areas of the brain. Because glucose is the main source of energy in the nonstarvation state of the brain, a PET scan shows how well brain cells are using glucose and how the neurons are firing.
18F-FDG PET has been used for studies of metabolic patterns in different dementia disorders (19). 18F-FDG PET shows that in AD, metabolic deficits are present in the neocortical association areas, with sparing of the basal ganglia, thalamus, cerebellum, primary sensory motor cortex, and visual cortex (20). Figure 1A illustrates a typical 18F-FDG PET scan of cerebral glucose metabolism in a patient with AD. PET of glucose metabolism in AD patients shows a consistent pattern of reduced glucose metabolism in the precuneus, posterior cingulate cortex, and parietotemporal regions and extending to the frontal cortex along with advancing disease (21,22). 15O-labeled H2O is a PET tracer used to study cerebral blood flow and has been used in dementia research. The regions of reduced cerebral blood flow in AD are the same as the regions of reduced regional CMRglc in AD (23). A limitation of the 15O-H2O PET method is that it can be performed only in a PET center with a cyclotron unit because of the short half-life of 15O (2 min).
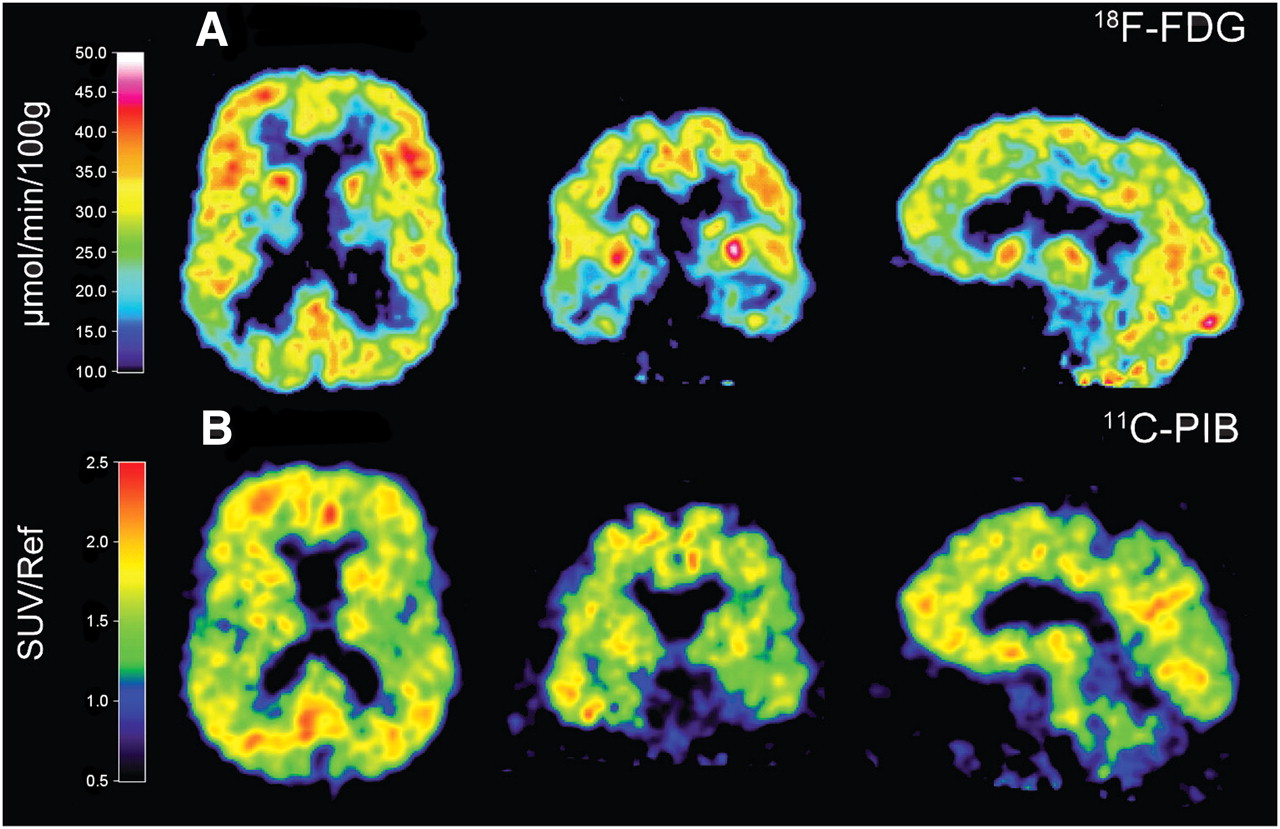
PET illustrating cerebral glucose metabolism as assessed by 18F-FDG, showing reduction of glucose metabolism in parietotemporal region (A) whereas amyloid plaque burden as assessed by 11C-PIB shows high PIB retention in large areas of brain (B) in 63-y-old patient with AD. 18F-FDG image was derived from Patlak analysis. 11C-PIB images were derived from summations of SUVs over 40–60 min and normalized with reference region cerebellum. Scale indicates high (red) to low (blue) tracer uptake or binding. Images represent horizontal, coronal, and sagittal sections.
Two brain regions, the anterior parahippocampal gyrus and the hippocampus, have shown significantly lower CMRglc in MCI patients than in healthy controls (24). In addition, CMRglc in the anterior parahippocampal gyrus correctly classified 85% of healthy controls and MCI patients (24). Reduction in CMRglc in the posterior cingulate cortex was observed in patients with early AD (25). In another study, hypometabolism of the posterior cingulate and temporoparietal cortex was observed in MCI patients, when compared with controls. Progression of some of these patients to probable AD showed additional bilateral hypometabolism in prefrontal areas, with further reduction in the posterior cingulate and parietal cortex, but no such changes in MCI patients who remained stable (26). In MCI patients, delayed episodic memory performance significantly correlated with hypometabolism in the frontal cortex (27). Comparison between the 2 MCI subtypes (amnestic and nonamnestic) showed temporal lobe hypometabolism in amnestic MCI subjects, possibly reflecting the conversion to AD (28,29). Glucose metabolism is a sensitive measure of change in cognition and functional ability both in AD and in MCI and has value in predicting future cognitive decline (30).
Longitudinal studies have demonstrated a significant reduction of CMRglc in the frontal, parietal, temporal, and posterior cingulate regions in AD subjects, which contrasts with the absence of significant decline in healthy control subjects (31). During disease progression, glucose metabolism continues to decline and is associated with worsening scores in cognitive tests (32). This observation indicates that 18F-FDG PET can serve as a biomarker in therapeutic trials. Indeed, several cholinesterase inhibitors such as donepezil (33), rivastigmine (34,35), galantamine (36), and phenserine (37) have shown positive effects on CMRglc in patients with mild to moderate AD.
Functional changes in the brain can be detected in subjects at risk of AD. Asymptomatic carriers of mutations of either the amyloid precursor protein gene or the presenilin gene have been shown to have progressive parietotemporal hypometabolism resembling the pattern seen in clinical AD (38,39). The apolipoprotein E (APOE) ε4 allele is a susceptibility gene that generally is overrepresented in patients with AD. Significantly lower glucose metabolism in AD-related brain regions has been reported in cognitively normal ε4 carriers than in non-ε4 allele carriers (40–42). Furthermore, a relationship between reduced CMRglc in AD-vulnerable brain regions and a maternal family history of AD in cognitively normal individuals has been observed (43).
Characteristic 18F-FDG PET metabolic patterns are observed in patients with other types of dementia. In DLB, a deficit in CMRglc has been observed in the temporal, parietal, occipital, and cerebral cortices (44,45). The hallucinations that are a clinical symptom of DLB are likely a correlate of a reduction of CMRglc in the primary visual cortex (46); in contrast, metabolic activity in the primary visual cortex is usually well preserved in AD. In another PET study, significantly lower CMRglc was revealed for the visual cortex (Brodmann areas 17–19) in the DLB group than the AD group, but no differences were found in other regions commonly affected in AD, including the posterior cingulate, superior parietal lobe, lateral temporal lobe, and prefrontal region (47). Delusions are frequent in DLB, and a significant relative hypometabolism of the right prefrontal cortex was found in the DLB patients with delusions (48). For differential diagnosis between AD and DLB, 123I-labeled N-ω-fluoropropyl-2β-carbomethoxy-3β-(4-iodophenyl)nortropane) (FP-CIT), reflecting dopamine uptake sites, might be preferable to 18F-FDG for discrimination (49). An improvement in diagnostic accuracy between DLB and AD, with a sensitivity of 78% and specificity of 94%, has been reported using 123I-FP-CIT (50).
Patients with FTD show hypometabolism mainly in the frontal, anterior temporal lobe (51). The regional pattern of predominantly frontal impairment usually allows clear distinction from AD, although there may be some overlap because AD can involve frontal regions and FTD may not spare the temporoparietal cortex (21). A series of 45 patients with a pathologically confirmed diagnosis showed that 18F-FDG PET can discriminate FTD from AD with more than 85% sensitivity and specificity (52). It might be difficult to discriminate between FTD and AD using 18F-FDG PET. Structural imaging adds value because, commonly, marked atrophy is observed in the frontal and anterior temporal lobe (often asymmetric) (6).
In PDD, reduced CMRglc has been observed in the parietal, frontal, lateral temporal, and visual cortices (53); however, PET of dopamine receptors is more specific for the disease.
Imaging of CMRglc with 18F-FDG PET is highly sensitive and specific for early differential diagnosis of the forms of dementia. In the United States, Medicare (the government-administered program of health insurance) covers 18F-FDG PET for the discrimination of AD from FTD.
IMAGING OF NEUROTRANSMITTER ACTIVITY
PET tracer studies on neurotransmitter systems in dementias may increase the understanding of the pathophysiologic mechanisms of different disorders and thereby improve diagnostic accuracy, because in certain diseases a particular neurotransmitter system is predominantly involved. The major transmitter systems that are impaired in dementias are the cholinergic, serotonergic, dopaminergic, glutaminergic, and noradrenergic systems. AD is characterized by impairment of the neocortical cholinergic system (54), whereas in PD and related dementias, major disturbances exist in the dopaminergic systems but the cholinergic system may also be impaired (55). Less is known about neurotransmitter changes in FTD, but a selective vulnerability of serotonergic neurons was recently observed (56). The PET tracers that have been developed for neurotransmitter systems and applied to studies of patients with various dementias are discussed in this section.
Cholinergic Neurotransmission
Acetylcholinesterase Activity.
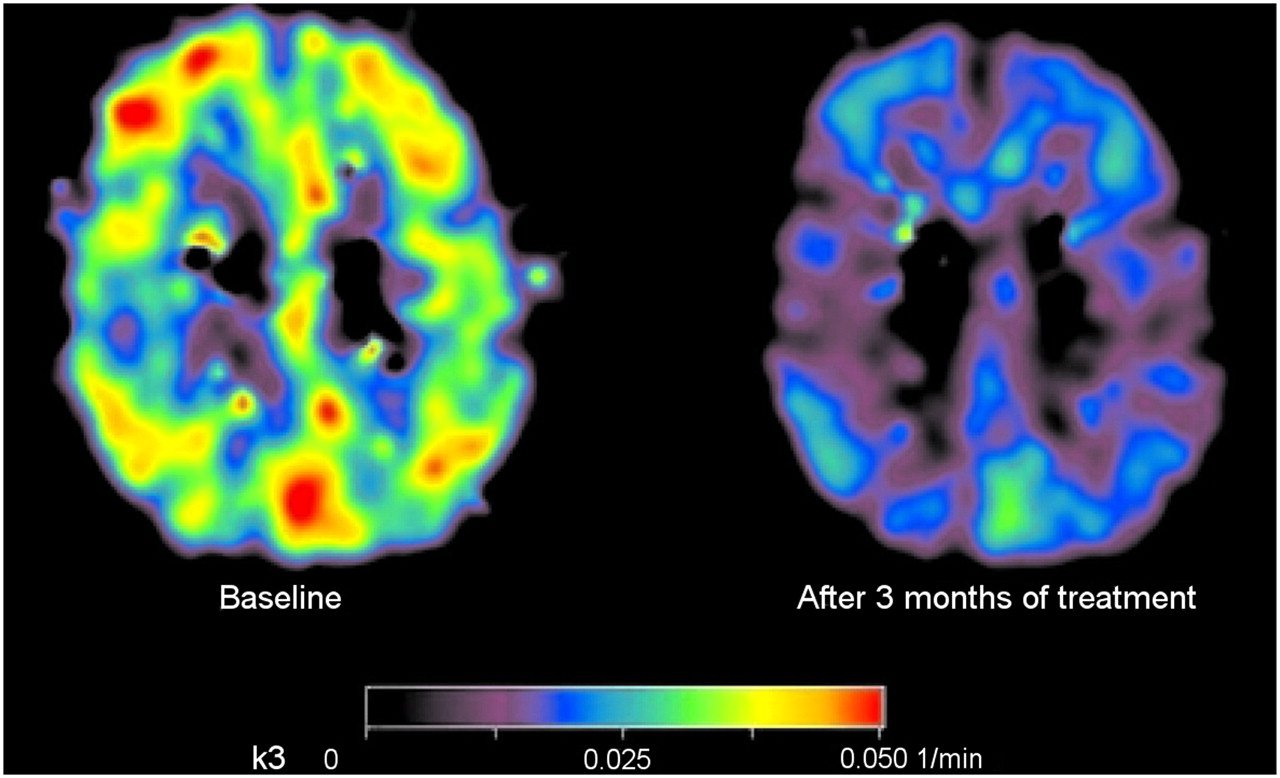
The most important degrading enzyme for acetylcholine in the human cortex is acetylcholinesterase, which is present in cholinergic axons. In the cortex, acetylcholinesterase is present in axons innervating it from the basal forebrain (57). There also is acetylcholinesterase in intrinsic cortical neurons, and low levels of acetylcholinesterase are probably present in the noncholinergic structures postsynaptic to the nucleus basalis innervation (58). As the cholinergic axons degenerate, acetylcholinesterase activity is reduced. The labeled analogs of acetylcholine that are substrates for acetylcholinesterase can be used for measuring and imaging its activity in vivo. Thus, the PET tracers N-11C-methylpiperidine-4-yl propionate (11C-PMP) and N-11C-methylpiperidyl-4-yl acetate (11C-MP4A) have been used to measure acetylcholinesterase activity (59,60), and a decline was found in AD patients, compared with healthy controls (21). Eggers et al. (61) studied 11C-MP4A imaging in AD patients with different APOE ε4 genotypes and found that acetylcholinesterase activity was lower in AD patients who were not APOE ε4 allele carriers than in those who had the APOE ε4 allele. These results indicate that cortical acetylcholinesterase activity is relatively well preserved in APOE ε4 carriers, either by preservation of its cellular expression or as acetylcholinesterase activity in amyloid plaques. A significant correlation between attention tests such as digit span and cortical acetylcholinesterase activity, measured by 11C-PMP, has been described in AD patients (62). Reduction in acetylcholinesterase activity has also been reported in MCI patients (63), especially in MCI patients who later convert to AD (64), suggesting that acetylcholinesterase changes might precede the development of clinical AD. The PET technique has also been used to measure drug-induced acetylcholinesterase inhibition in AD patients, which for all currently available cholinesterase inhibitors at the standard clinical dose ranges from 30% to 40% (65–67). Figure 2 illustrates the cortical acetylcholinesterase inhibition measured by 11C-PMP in a patient with mild AD after 3 mo of treatment with cholinesterase inhibitor galantamine, compared with baseline.
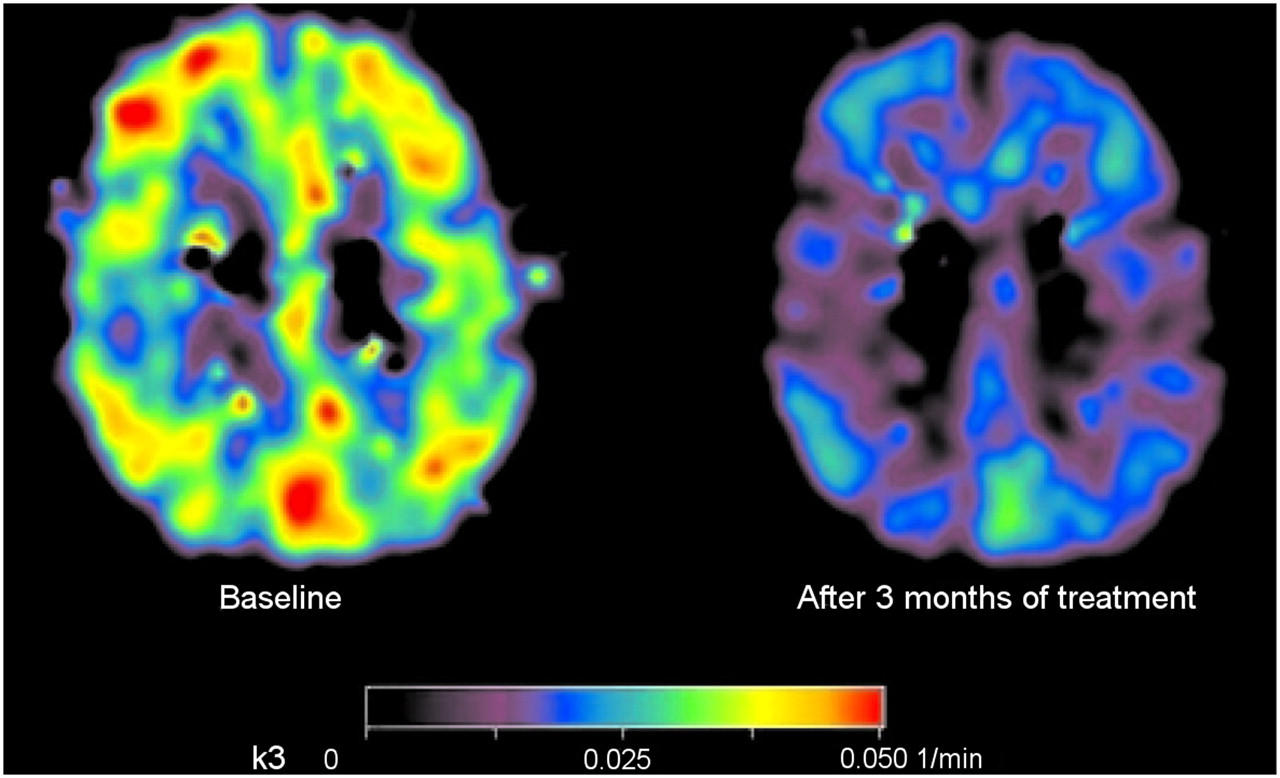
Parametric map illustrating regional cortical acetylcholinesterase activity (k3) before (left) and after 3 mo (right) of galantamine treatment in patient with mild AD measured using 11C-PMP. (Reprinted with permission of (67).)
Patients with PDD have lower acetylcholinesterase activity than do AD patients (55), suggesting more severe cholinergic pathway deficits. Cortical acetylcholinesterase activity measured by 11C-PMP is associated with cognitive test attention in PDD patients, as in AD patients (68). Patients with DLB also show a decrease in cerebral acetylcholinesterase activity as measured by 11C-MP4A (69).
Cholinergic Receptors.
The main receptors for acetylcholine in the central nervous system are muscarinic and nicotinic receptors of various subtypes. The nicotinic acetylcholine receptors (nAChRs) are ligand-gated ion channels comprising 5 subunits, variously consisting of α2–α10 and β2–β4. The nAChRs have been implicated in a variety of central processes, such as attention, memory, and cognition. There are 3 general classes of compounds developed as PET radiotracers for nAChRs: nicotine and its derivatives, epibatidine or azetidine and their derivatives, and 3-pyridyl ether derivatives, including A-85380 (70).
PET studies using (S)(−)-11C-methyl nicotine (11C-nicotine) have shown a decrease in the number of cortical nAChRs in AD patients (71). Decreased cortical 11C-nicotine binding in AD patients significantly correlates with impaired cognitive function, such as impaired attention (72). When 11C-nicotine binding was measured by PET before and after treatment with cholinesterase inhibitors in mild-AD patients, an increased number of cortical nicotinic receptors was observed after cholinesterase inhibitor treatment. This increase significantly correlated with improvement in attention in the AD patients (67,73).
The PET tracer 2-18F-fluoro-A-85380 has been developed to visualize a specific subtype of nAChRs, that is, α4β2. The major drawback with 2-18F-fluoro-A-85380 is the considerably longer scanning time (7–8 h) it requires. Fewer clinical studies have been done with 2-18F-fluoro-A-85380. Ellis et al. did not observe any significant loss of in vivo cortical nAChRs in patients with AD, compared with age-matched healthy controls (74); however, using the same tracer, Sabri et al. showed a reduction of α4β2 receptors in both AD and MCI patients (75). In vivo reductions of α4β2 nAChR binding in the striatum and substantia nigra have also been reported in PD without dementia (76). Because of the extraordinarily long acquisition time required for 2-18F-fluoro-A-85380, the new α4β2 nAChR-specific radioligands (+)- and (−)-18F-norochloro-fluoro-homoepibatidine have been evaluated preclinically in the porcine brain and shown 2-fold higher brain uptake and significantly shorter acquisition times (77). However, SPECT studies using 123I-5IA-85380 have shown a reduction of nAChRs in several cortical brain areas in AD patients (78). In patients with PD, compared with healthy volunteers, widespread reduced 2-18F-fluoro-A-85380 binding was observed, especially in the midbrain, pons, cerebellum, and several cortical areas. In addition, patients with PD and associated depression showed a pronounced reduction of 2-18F-fluoro-A-85380 binding, mostly in the cingulate cortex and frontoparietooccipital cortex (79). An interesting approach is to develop PET tracers selective for α7 nAChRs, because this receptor subtype may have a broad interaction with several neurotransmitter systems and pathologic processes in AD (80). Recently, the results of PET studies have suggested that 11C-CHIBA-1001 may be a suitable radioligand for imaging α7 nAChRs in the human brain, providing acceptable dosimetry and pharmacologic safety at a dose required for adequate PET images (81).
Fewer PET studies have been performed to examine the muscarinic receptors in AD. Zubieta et al. studied the nonselective muscarinic receptor ligand 11C-N-methyl-4-piperidyl benzilate and did not observe any changes in AD patients (82). 18F-fluoropropyl-thiadiazolyltetrahydro-1-methylpyridine, a selective tracer for M2 receptor, showed high binding in subjects who were APOE ε4 carriers (83).
Serotonergic Neurotransmission
Postmortem studies have shown a widespread loss of serotonin (5-hydroxytryptamine [5-HT]) subtypes 1 (5-HT1) and 2 (5-HT2) receptors in the amygdala and hippocampus of AD brains. In vivo PET studies have also demonstrated a loss of some subtypes of 5-HT receptors in the AD brain. 18F-altanserin, a tracer for 5-HT2A receptor, showed a 40% reduction in binding potential in AD patients, particularly in the amygdala–hippocampal complex, anterior cingulate, prefrontal cortex, lateral temporal cortex, and sensorimotor cortex, whereas a somewhat smaller loss was observed in the parietal cortex, compared with the age-matched healthy controls (84). A single study by Kristiansen et al. (85) quantified cerebral 5-HT2A receptor density in FTD patients, using 11C-MDL, a selective tracer for 5-HT2 receptor and PET. The in vivo measurements indicated a significant reduction in 5-HT2 receptor in the orbitofrontal, frontal medial, and cingulate cortices in FTD.
Hippocampal pyramidal neurons show a high concentration of 5-HT1A receptor, and loss of these pyramidal neurons has been correlated with lower 5-HT1A receptor densities in AD brains (86). More recent PET studies therefore have focused on changes in 5-HT1A receptor in AD. PET studies using 18F-2′-methoxyphenyl-(N-2′-pyridinyl)-p-fluoro-benzamidoethyipiperazine (MPPF), a selective ligand for 5-HT1A receptor, showed diminished hippocampal binding in AD patients, whereas patients with MCI showed binding values intermediate between controls and patients with AD (86). The differential binding potential of 18F-MPPF in the hippocampus might indicate neuronal losses and may eventually prove valuable as an early diagnostic measure even before symptoms are present, but further PET studies are needed to confirm this possibility.
Dopaminergic Neurotransmission
PET makes it possible to study aspects of dopaminergic neurotransmission such as synthesis, storage, receptor binding (D1 and D2), reuptake, and the vesicular transporter of dopamine.
The most widely used tracer to examine dopamine synthesis and vesicular storage is 6-18F-fluoro-l-dopa. A decrease in uptake of striatal 6-18F-fluoro-l-dopa was observed in PD and PDD, without differences between groups (87). A deficit of dopamine synthesis similar to that in PD has been found in DLB, even at a stage when parkinsonism may not yet be prominent (88), whereas no similar abnormality is seen in patients with AD.
Compared with healthy controls, AD patients have shown a reduction in striatal dopamine D1 receptors as measured by 11C-NNC756 PET but not in dopamine D2 as measured by 11C-raclopride PET (89). However, striatal D2 receptor density, measured by 11C-raclopride, has been shown to be associated with severe behavioral symptoms in AD (90). Extrastriatal dopamine D2 receptors evaluated by 11C-labeled (S)-N-((1-ethyl-2-pyrrolidinyl)methyl)-5-bromo-2,3-dimethoxybenzamide (FLB 457) have shown reduced binding potential in the hippocampus and temporal cortex associated with changes in memory and language performance in AD patients (91).
11C-raclopride, a well-established D2 receptor antagonist, has been used to detect signs of nigrostriatal degeneration by PET. An increase in D2 receptor binding was observed in patients with early PD (92), and a reduction in raclopride binding was observed after levodopa treatment (93). Neuroimaging of dyskinesia has been performed using 11C-labeled R-(+)-8-chloro-2,3,4,5-tetrahydro-3-methyl-5-phenyl-1H-3-benzazepine-7-ol) (SCH-23390) (D1 receptors) and 11C-raclopride (D2 receptors) (94). No significant reduction in mean 11C-raclopride uptake was observed in either the putamen or the caudate nucleus in AD patients (89).
Striatal dopamine reuptake sites have been studied with PET in AD patients using the cocaine analog 11C-labeled (−)-2β-carbomethoxy-3β-(4-fluorophenyl)tropane (β-CFT) as a radioligand. Compared with control subjects, AD patients showed a reduction in 11C-β-CFT uptake in the putamen and caudate nucleus (95). In PD, a decrease in 18F-β-CFT uptake was observed in the putamen and caudate nucleus (96). 123I-FP-CIT is used in SPECT for measuring dopamine uptake sites and discriminates between AD and DLB (50).
(+)-11C-dihydrotetrabenazine (11C-DTBZ), a ligand that binds to monoamine transporter and is less regulated by intrasynaptic dopamine than are other dopaminergic tracers, is used as a reliable marker of dopaminergic presynaptic terminal integrity. 11C-DTBZ PET can be used to differentiate DLB from both PD and AD (97).
PATHOLOGY MARKERS
Recent developments in molecular imaging have opened up the possibility of imaging pathologic processes involving amyloid proteins, τ-protein, activated microglia, and astrocytes.
Amyloid Imaging
Amyloid imaging in AD has developed rapidly in recent years. Because Aβ plaque is the hallmark of AD pathology, much effort has gone into developing radiotracers that allow imaging of Aβ plaques in vivo (98). The small-molecule approach led to different compounds emanating from Congo red, chrysamine-G, and thioflavine to be used in amyloid imaging for PET (98). An important clinical question is whether amyloid imaging can differentiate AD from other forms of dementia.
18F-FDDNP.
2-(1-{6-[(2-18F-fluoroethyl)(methyl)amino]-2-naphthyl}ethylidene)malononitrile (18F-FDDNP) was the first PET tracer used in vivo for cerebral amyloid plaque (99). Increased 18F-FDDNP binding was observed in the temporal, parietal, and frontal regions of the AD brain, compared with older control subjects without cognitive impairment (99).
18F-FDDNP binding in MCI patients has been reported to be intermediate between controls and AD patients (100). In addition, 18F-FDDNP has been shown to bind to τ-protein in AD (100), and 18F-FDDNP signal in the medial temporal lobe has been shown to be associated mainly with τ-pathology whereas that in other areas of the brain is overwhelmingly related to amyloid plaque deposition (100). It has also been shown that 18F-FDDNP binds to protease-resistant prion protein in an amyloid form in postmortem tissues in Creutzfeldt–Jacob disease (101) and in vivo in a patient with prion disease (102). A negative correlation between 18F-FDDNP binding and CSF Aβ levels and positive correlations with CSF τ-level have been observed in AD and MCI patients (103).
11C-PIB.
Pittsburgh compound B (11C-PIB), a derivative of thioflavin-T amyloid dye, binds with high affinity and high specificity to neuritic Aβ plaques (104) but not at all to diffuse plaque and neurofibrillary tangles (105). The first 11C-PIB studies on humans were in Sweden in 2002. Compared with healthy controls, 16 mild-AD patients undergoing imaging with 11C-PIB showed significantly higher (×1.9–1.5) 11C-PIB retention in the frontal, temporal, parietal, and occipital cortices and the striatum (106). Retention of 11C-PIB was low and similar in the pons and cerebellum of both groups (106). For 11C-PIB, dynamic scanning quantifies binding potentials, and a relatively simple and practical way of quantifying uptake in a clinical setting is based on scans obtained approximately 60–90 min after intravenous injection of the tracer, with the cerebellum as the unaffected reference region (107). Figure 1 illustrates the high 11C-PIB retention in large areas of the brain, compared with the more regional selective decrease in 18F-FDG uptake in a patient with mild AD. 11C-PIB has now been used by a large number of PET centers all over the world and consistently shows high sensitivity in detecting amyloid plaques and vascular amyloid protein in vivo (22,108). In a recent study, patients undergoing brain biopsy and showing high Aβ aggregation in the frontal cortical biopsy specimen also revealed high cortical 11C-PIB uptake with PET (109). Furthermore, a significant correlation has been reported between 11C-PIB imaging in vivo and levels of Aβ in postmortem brain tissue (110). The strong correlation between in vivo 11C-PIB retention and region-matched quantitative analysis of Aβ plaques at autopsy supports the validity of 11C-PIB PET as a method for in vivo evaluation of Aβ plaque burden (110).
When AD patients underwent both 11C-PIB and 18F-FDDNP PET, lower binding was observed with 18F-FDDNP than with 11C-PIB (111), and 18F-FDDNP showed high binding in the medial temporal lobe, probably because FDDNP binds to neurofibrillary tangles (111). Autoradiographic studies on postmortem brain tissue also confirmed lower binding of 3H-FDDNP than of 3H-PIB to AD brain tissue—10 times lower affinity for 3H-FDDNP than for 3H-PIB to fibrillar amyloid deposition (112).
Other Amyloid PET Ligands.
Several other PET ligands have also recently been tested in healthy subjects and AD patients. Table 2 lists the different 11C- and 18F-compounds that have been explored so far for amyloid imaging in demented patients. Because the half-life of 18F is longer than that of 11C, 18F-compounds are promising for routine use in a clinical setting. When comparing 18F-florbetaben (18F-BAY-94-9172) with 11C-PIB, Rowe et al. (113) concluded that 11C-PIB seems to show a greater difference between AD patients and controls than does 18F-BAY-94-9172. 11C-SV13 showed a binding potential similar to that of 11C-PIB in a small sample of AD patients (114). 11C-2-(2-[2-dimethylaminothiazol-5-yl]ethenyl)-6-(2-[fluoro]ethoxy)benzoxazole (11C-BF-227) was suggested to be more sensitive for detecting amyloid plaque at later stages of AD disease (115), but in a recent study 11C-BF-227 detected amyloid pathology in MCI patients (116). 11C-labeled 5-(6-{[tert-butyl(dimethyl)silyl]oxy}-1,3-benzothiazol-2-yl)pyridin-2-amine (1C-AZD-2184) (117), 18F-flutemetamol (118), and (E)-4-(2-(6-(2-(2-(2-18F-fluoroethoxy)ethoxy)ethoxy)pyridin-3-yl)vinyl)-N-methyl benzeneamine (18F-AV-45) (119,120) are new amyloid PET ligands that have been tested in AD patients.
Time Course of Amyloid and Functional Changes in AD Measured by PET.
Autopsy studies on the AD brain have shown that Aβ deposition reaches a ceiling early in the disease process and remains stable during disease progression, whereas neurofibrillary tangle formation, synaptic loss, and gliosis continue throughout the course of the illness (121). A crucial question has been whether PET amyloid imaging shows changes in amyloid load in the brain as AD progresses. In a 2-y follow-up study on 16 AD patients, 11C-PIB retention had not changed between baseline and the 2-y follow-up scan, suggesting an apparent plateau in amyloid load despite a decline of cerebral glucose metabolism and cognition (32). The unchanged 11C-PIB retention after 2 y of follow-up may suggest a different time course for amyloid load than for changes in the functional activity in the brain (122). It is possible that the amyloid load maximizes in almost the prodromal stage of AD, as is also supported by 11C-PIB PET studies on MCI patients (123). These observations are supported by other recent 1- and 2-y follow-up studies showing no significant change in 11C-PIB retention in AD patients although brain atrophy progressed significantly (124,125). Even after 5 y of follow-up, no changes in 11C-PIB retention were observed at the group level in AD patients (126).
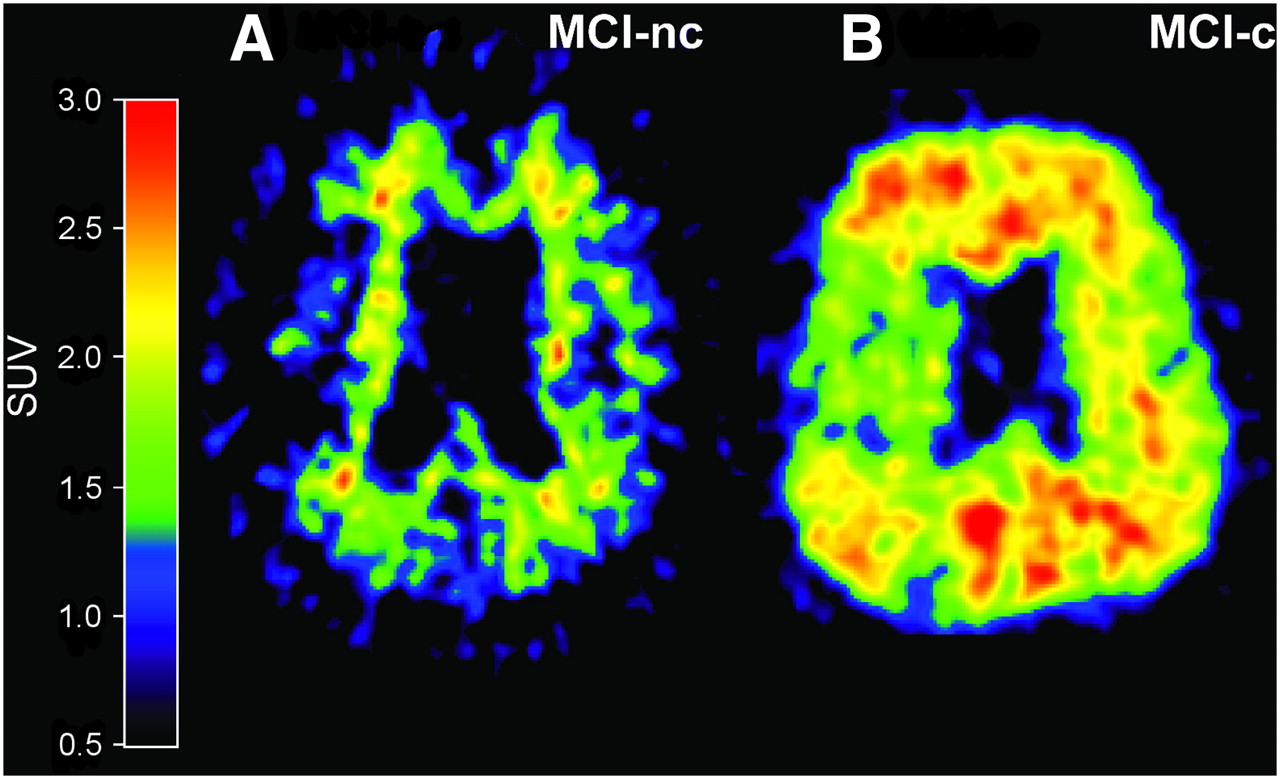
Studies on 11C-PIB retention in MCI patients suggest that the amount of PIB binding is bimodally distributed, with one subset of MCI patients showing abundant neocortical binding (PIB-positive) and another subset showing only low, nonspecific binding (PIB-negative), indicating a lack of fibrillar amyloid deposition (127–129). Clinical follow-up studies have shown that MCI patients who converted to AD showed significantly higher 11C-PIB retention than did MCI nonconverters, suggesting the possibility of identifying prodromal AD by amyloid imaging (123,130,131). Figure 3 shows 11C-PIB images of MCI converters and nonconverters to AD.
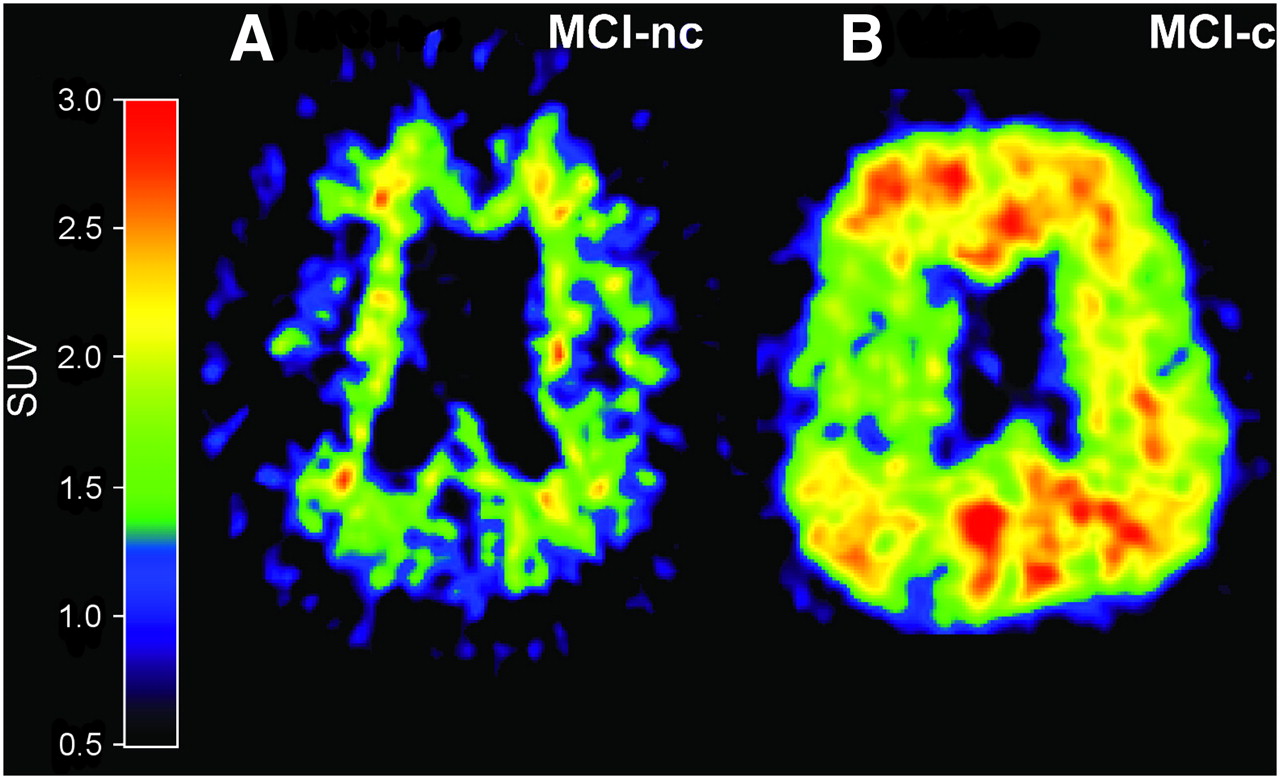
In vivo amyloid imaging as assessed by 11C-PIB in 2 MCI patients, one with low PIB binding (PIB-negative) who during follow-up period did not convert to AD (MCI-nc) (A) and another with high PIB binding (PIB-positive) who later during clinical follow-up converted to AD (MCI-c) (B).
An interesting question is how early a deposit of amyloid plaque can be detected in prodromal AD. Studies of amyloid imaging in subjects at risk for developing AD will be of great importance. An early high PIB retention in the striatum was reported in presymptomatic carriers of presenilin-1 mutation (132). Similarly, an increased PIB uptake also in the striatum and posterior cingulate was observed in patients with amyloid precursor protein locus duplication and cerebral amyloid angiopathy (133). Compared with a sporadic-AD patient, a familial-AD patient who carried His163Tyr mutation in the presenilin-1 gene showed high PIB retention in the striatum but also in the posterior cingulate cortex and the thalamus (39). Variant-AD patients with certain mutations of the presenilin-1 gene showed high PIB retention in the striatum (caudate nucleus and putamen), anterior and posterior cingulate gyrus, occipital cortex, and thalamus (134).
Although 11C-PIB seems to be more accurate than 18F-FDG PET in discriminating AD (accuracies of 90% and 70%, respectively) (135), a less significant correlation has been observed between episodic memory and 11C-PIB retention in the brains of AD and MCI patients (32,123,136,137). The stronger correlation between cognitive performance and cerebral glucose metabolism in AD patients suggests that the decline in glucose metabolism parallels the decline in cognition during disease progression, reflecting neurodegeneration (22,138). A significant negative correlation has been observed between CSF Aβ42 and cortical 11C-PIB retention in AD and MCI patients (139,141), suggesting that PET amyloid imaging and CSF Aβ42 appear to be earlier markers for prodromal AD than are cerebral glucose metabolism and cognitive testing.
Interestingly enough, high 11C-PIB retention has been observed in healthy elderly control subjects (106,142,143). Some 11C-PIB–positive elderly healthy controls have demonstrated impairment on cognitive tests (137), whereas other 11C-PIB–positive healthy controls have shown normal cognition (144). Longitudinal studies are needed to find out if these aged healthy subjects are in a prodromal stage of AD with pathologic formation of Aβ or whether the high amyloid load in older brains is due to slower clearance of amyloid plaque from the brain. A large longitudinal Australian study estimated that 25% of elderly individuals more than 85 y old showed a high amyloid load in the brain (145). A group of older adults who had no dementia and were PIB-positive showed greater declines in regional cerebral blood flow in certain brain areas than did PIB-negative healthy subjects—probably a sign of decrements in neuronal function (146). In a recent longitudinal study of 159 participants, Morris et al. reported that subjects with normal cognition at baseline and 11C-PIB–positive PET showed a greatly increased risk of developing symptomatic AD in the next 2–5 y, compared with PIB-negative subjects (147).
Amyloid Imaging in FTD, PDD, and DLB.
Amyloid plaque imaging might be important in diagnosing AD early and differentiating it from other forms of dementia. Generally, low cortical 11C-PIB retention was observed in patients with FTD (129,148), as well as in PD patients (129,149,150). Figures 4C and 4D illustrate low PIB retention in a PD patient without dementia and a patient with FTD. However, elevated 11C-PIB retention in the pons and mesencephalon was observed in PDD patients, compared with healthy controls (150). These findings suggest that 11C-PIB PET can be used to differentiate FTD, PD, and PDD from AD with respect to cortical amyloid retention.

High PIB retention was observed in patient with AD (A) and patient with DLB (B). In contrast, low PIB retention was observed in patient with PD (not demented) (C) and in patient with FTD (D). (Part C reprinted with permission of (149); part D courtesy of Professor Henry Engler, Uppsala PET Center, Academical Hospital, Uppsala, Sweden.)
Patients with DLB have often shown high 11C-PIB retention in the brain (Fig. 4B) (129,151,152), thought to be caused by binding to amyloid plaque and not to synuclein (153). Whether a combination of amyloid plaque and Lewy body pathology exacerbates the memory and cognitive problems in DLB is unclear, but such a combination is likely to accelerate the dementia process. It is plausible that amyloid imaging can be used to differentiate amyloid-positive DLB from amyloid-negative forms of DLB (154). Occipital PIB retention has been shown to be preferentially higher in DLB patients than in AD patients (152), and occipital hypometabolism in DLB may therefore be a consequence of this relatively higher occipital amyloid burden. At autopsy, a PIB-positive DLB patient showed PIB binding to vascular amyloid depositions (155).
High retention of this tracer has also been observed in patients with cerebral amyloid angiopathy, although the regional distribution of 11C-PIB in the brain seems to be somewhat different between this condition and AD (156).
Amyloid Imaging and Drug Evaluation.
Different treatment strategies to reduce the amyloid load in the brain of AD patients are being tested. Active and passive immunotherapy is a focus of investigation. In vivo imaging of amyloid plaque in patients undergoing antiamyloid treatment has great potential for several reasons. In vivo 11C-PIB imaging before the start of treatment would allow selection of patients with a demonstrably high amyloid load in the brain. Repeated studies of 11C-PIB retention during ongoing treatment would allow detection of a decrease in insoluble Aβ load in the brain. In a recent study, phenserine, which inhibits formation of β-amyloid precursor protein, showed a reduction of Aβ load in AD brain that correlated negatively with CSF amyloid levels (37). Ongoing studies of immunization therapies are measuring 11C-PIB retention both before and during immunization. These studies will improve our understanding of the underlying mechanisms of antiamyloid therapy.
Microglia
The process of neurodegeneration in AD is associated with a local glial response within the brain parenchyma that involves activation of microglia. Activated microglia have a key role in the immune response of the brain to neuronal degeneration. Activated microglia are present at sites of aggregated Aβ deposition in the brains of AD subjects. Quantitative in vivo measurements of glial activation have used 1-[2-chlorophenyl]-N-methyl-N-[1-methylpropyl]-3-isoquinoline carboxamide (11C-(R)-PK11195), a specific ligand for the peripheral benzodiazepine binding receptor site. PET studies showed an increased in 11C-(R)-PK11195 binding in the entorhinal, temporoparietal, and cingulate cortices in patients with mild AD, compared with healthy control subjects (157). In a recent study, Edison et al. demonstrated high microglial activation by 11C-(R)-PK11195 in a group of AD subjects with high PIB retention. A negative correlation was reported between levels of cortical microglial activation and cognition, whereas no correlation was observed between PIB binding and 11C-(R)-PK11195 (158). A new approach to modeling the signal from 11C-(R)-PK11195 binding might, however, be needed to increase the sensitivity of the tracer, and a recent study showed that inclusion of a vascular component amplifies the binding potential in AD patients (159). PET scans with 11C-labeled N-(2,5-dimethoxybenzyl)-N-(5-fluoro-2-phenoxyphenyl)-acetamide (11C-DAA1106), a new and selective ligand for peripheral benzodiazepine binding receptor sites, have been performed on patients with AD and showed high binding potential in the AD brain, compared with controls (160).
Patients with PD have shown significantly increased mean levels of 11C-(R)-PK11195 binding in the pons, basal ganglia, and frontal and temporal cortical regions, compared with control subjects (161). Levels of microglial activation did not correlate with clinical severity or putaminal 6-18F-fluoro-l-dopa uptake (161), indicating that widespread microglial activation is associated with the pathologic process in PD. Parallel changes in microglial activation by 11C-(R)-PK11195 and corresponding dopaminergic terminal loss by 11C-CFT have also been demonstrated in the affected nigrostriatal pathway in early PD (162). Elevated levels of 11C-(R)-PK11195 have been observed in brains of patients with FTD (163) and Huntington disease (164).
Astrocytes
Autopsy brain studies have shown an increased number of astrocytes in dementia disorders. N-[11C-methyl]-l-deuterodeprenyl (11C-DED) binds irreversibly to the enzyme monoaminooxydase-B, which is expressed by reactive astrocytes and can be used as a marker of reactive astrocytosis. A study of patients with Creutzfeldt–Jacob disease showed an increase in 11C-DED binding accompanied by a decrease in cerebral glucose metabolism (165). Further studies are needed to verify the use of 11C-DED in neurodegenerative disorders. PET studies with 11C-DED in AD and MCI patients are in progress.
CONCLUSION
Clinical, pathologic, and genetic evidence indicates that the neurodegenerative dementias have different underlying etiologies and pathogenetic mechanisms. PET allows the measurement of pathologic processes such as those involving amyloid plaque, reactive microglia, and astrocytes, as well as neurodegeneration reflected by declining cerebral glucose metabolism and impaired neurotransmitters. Accurate and early differentiation of dementias will become increasingly important as new therapies are introduced. Differential diagnosis by standard clinical criteria has limited accuracy. However, the rapid development of the field of molecular imaging is presently promising. PET offers the potential to increase diagnostic accuracy. Treatment approaches probably will be different for each major form of degenerative dementia.
Recently, reliable PET tracers for assessing the Aβ burden in the brain have been introduced, and they may have high value for early diagnosis at presymptomatic stages of AD and more accurate differential diagnosis from other forms of dementia. Amyloid imaging can detect the presence of amyloid plaque in the brain at prodromal stages of AD, whereas the cerebral glucose metabolism and decline in cognition more closely follow the clinical symptoms and progression of AD. For evaluating the effect of new disease-modifying therapeutics, the combined use of brain glucose metabolism, amyloid, and microglial imaging should allow us to determine whether antiamyloid strategies clear plaques and exert therapeutic effects via stimulating or suppressing glial activation, as well as ceasing neuronal loss. It will be important to combine findings from PET with other diagnostic markers such as MRI, CSF biomarkers, and cognitive measurements to increase diagnostic accuracy.
Acknowledgments
The financial support of the Swedish Research Council (project 05817), Stohne's Foundation, Foundation of Old Servants, KI Foundations, Alzheimer Foundation in Sweden, Demensfonden, Brain Foundation, Swedish Brain Power, Knut and Alice Wallenberg Foundation, and EC-FP6-project DiMI, LSHB-CT-2005-512146, is highly acknowledged. Anders Johansson and Henry Engler are thanked for providing the 11C-PIB PET images for PD and FTD.
Footnotes
Learning Objectives: On successful completion of this activity, participants should be able to describe (1) different PET tracers for measuring functional activity, neurotransmitter systems, and pathologic markers in the brain of patients with dementia; (2) how to use different PET tracers to study the typical pattern of changes in different types of dementia; and (3) how to discriminate between the different dementia disorders using PET in clinical practice.
Financial Disclosure: The authors of this article have indicated that they serve as board members, officers, or trustees of Elan, Pfizer, GlaxoSmithKline, Novartis, Lundbeck Inc., and GE Healthcare; are consultants or advisors for AstraZeneca; are meeting participants or lecturers for Novartis and Eli Lilly; and are involved with scientific studies or trials by Novartis, GlaxoSmithKline, Wyeth, and TorryPines Therapeutics, Inc. No other potential conflict of interest relevant to this article was reported.
CME Credit: SNM is accredited by the Accreditation Council for Continuing Medical Education (ACCME) to sponsor continuing education for physicians. SNM designates each JNM continuing education article for a maximum of 1.0 AMA PRA Category 1 Credit. Physicians should claim only credit commensurate with the extent of their participation in the activity.
For CE credit, participants can access this activity through the SNM Web site (http://www.snm.org/ce_online) through September 2011.
- © 2010 by Society of Nuclear Medicine
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.↵
- 124.↵
- 125.↵
- 126.↵
- 127.↵
- 128.
- 129.↵
- 130.↵
- 131.↵
- 132.↵
- 133.↵
- 134.↵
- 135.↵
- 136.↵
- 137.↵
- 138.↵
- 139.↵
- 140.
- 141.↵
- 142.↵
- 143.↵
- 144.↵
- 145.↵
- 146.↵
- 147.↵
- 148.↵
- 149.↵
- 150.↵
- 151.↵
- 152.↵
- 153.↵
- 154.↵
- 155.↵
- 156.↵
- 157.↵
- 158.↵
- 159.↵
- 160.↵
- 161.↵
- 162.↵
- 163.↵
- 164.↵
- 165.↵
- Received for publication November 4, 2008.
- Accepted for publication April 20, 2010.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.