


Abstract
Because of the recent development of an iodopyridopyrimidinone Abl protein kinase inhibitor (PKI), 124I-SKI-212230 (124I-SKI230), we investigated the feasibility of a PET-based molecular imaging method for the direct visualization of Abl kinase expression and PKI treatment. Methods: In vitro pharmacokinetic properties, including specific and nonspecific binding of 124I-SKI230 to its Abl kinase target and interaction with other PKIs, were assessed in cell-free medium and chronic myelogenous leukemia (CML) cells overexpressing BCR-Abl (K562), in comparison with BT-474 cells that are low in Abl expression. In a xenograft tumor model, we assessed the in vivo pharmacokinetics of 124I-SKI230 using PET and postmortem tissue sampling. We also tested a paradigm of 124I-SKI230 PET after treatment of the animal with a dose of Abl-specific PKI for the monitoring of the tumor response. Results: In vitro studies confirmed that SKI230 binds to Abl kinase with nanomolar affinity, that selective uptake occurs in cell lines known to express Abl kinase, that RNAi knock-down supports specificity of cellular uptake due to Abl kinase, and that imatinib, an archetype Abl PKI, completely displaces SKI230. With SKI230, we obtained successful in vivo PET of Abl-expressing human tumors in a nude rat. We were also able to demonstrate evidence of substrate inhibition of in vivo radiotracer uptake in the xenograft tumor after treatment of the animal as a model of PKI treatment monitoring. Conclusion: These results support the hypothesis that molecular imaging using PET will be useful for the study of in vivo pharmacodynamics of Abl PKI molecular therapy in humans.
Protein kinase inhibitors (PKIs) are enzymes involved in the transfer of phosphate from adenosine triphosphate (ATP) to tyrosine residues on polypeptides and proteins. The broad family of PKIs that blocks signal transduction mechanisms has become a new arena for molecular imaging strategies. Through overexpression and mutation, the protein kinases may become human oncogenes, such as endothelial growth factor receptor (EGFR) (lung cancer), HER2 (breast cancer), and BCR-Abl kinase (chronic myelogenous leukemia [CML]) (1–3). Several new drugs have been developed that are effective at tumor inhibition, largely through a strategy of empiric high-throughput screening of thousands of compounds against multiple distinct kinase targets (4–6).
The Philadelphia chromosome (Ph+) is present in 95% of CML and is formed as a result of a t(9,22)(q34;q11) translocation (7) producing a fusion oncogene, BCR-Abl, encoding for a constitutively activated protein tyrosine Abl kinase that is critical for the immortalization and malignant transformation of Ph+ CML (8). Ph+ activates the antiapoptotic mechanism through Hck and Stat5b signaling pathways (9) and is overexpressed in CML (10) and a subset of aggressive acute lymphoblastic leukemia (11). Ovarian cancer, squamous cell carcinomas, and other solid tumors may overexpress Abl (12–15).
PKIs of Abl, particularly imatinib mesylate (STI-571, Gleevec [Novartis], and CGP57148) (16), have provided major clinical benefit in treating CML. To overcome potential treatment failures due to resistance to these PKIs (17), a patient selection strategy may be used. Imatinib targets the ATP binding site of the Abl kinase domain, providing for selectivity and specificity. This fact has prompted us to search for PKI analogs targeting Abl kinase, in a manner similar to that of imatinib, to be radiolabeled to follow the pharmacokinetics of the drug in vivo using a direct imaging approach. These analogs are a potential aid in selecting patients for treatment and for detecting the emergence of resistance.
The direct imaging systems with radiolabeled PKIs have been successfully demonstrated for some other tyrosine kinases important in cancer-related signal transduction pathways. EGFR protein kinase inhibitors were imaged using PET (18,19). Radiolabeling of the clinically proved Abl inhibitors imatinib (20) and dasatinib (21) also demonstrated feasibility of PET in the murine xenograft models of CML.
To improve sensitivity of the direct imaging probe, we selected a series of PKI analogs of the pyridopyrimidinone class (22) with known high affinity, which shared similarities to Abl kinase binding with imatinib based on crystal structure (23). Representative compounds were made and radiolabeled, gaining 124I- and 131I-2-(4-iodophenylamino)pyrido[2,3-d]pyrimidin-7-one (SKI230; Fig. 1A) (24). The previously described SKI230 (24), with dual specificity to Abl and Scr ATP binding pockets, had an inhibitory effect on Abl kinase and cellular growth of a BCR-Abl–dependent cell line.

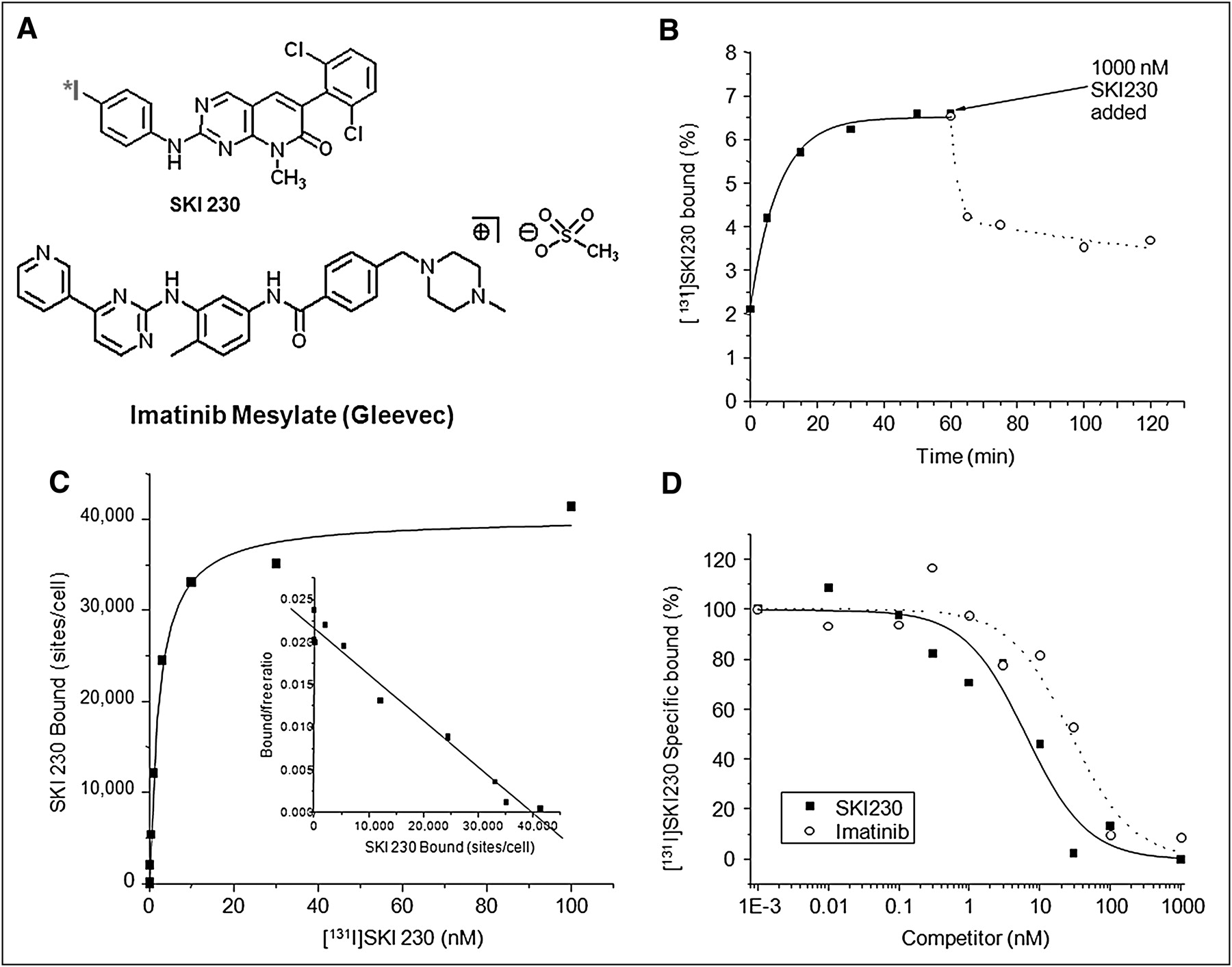
Pharmacochemical properties of Abl inhibitor for direct imaging. (A) Chemical structure of 124I-SKI230. (B) Time course of binding of 131I-SKI230 to K562 cells (solid line and ▪ = on rate; dotted line and ○ = off rate). (C) Saturation binding of SKI230 to K562 cells (inset Scatchard transformation). (D) Displacement binding of 131I-SKI230 to K562 cells by SKI230 (solid line and ▪) and imatinib (dotted line and ○).
In the current study, we assessed in vitro the radiolabeled SKI230 target binding and uptake in the cell lines with different levels of expression of Abl kinase. In vivo, we evaluated the feasibility of the use of a radiolabeled SKI230 for direct imaging of Abl protein kinase expression and a strategy of in vivo uptake blocking imaging as potential clinically relevant applications of SKI230 in the monitoring of treatment with Abl-specific inhibitors.
MATERIALS AND METHODS
Synthesis and Radiolabeling
SKI230 was synthesized and radiolabeled with 124I or 131I as reported previously (24). The chemical and radiochemical purity of the tracer was assessed before use by both radio–thin-layer chromatography and high-performance liquid chromatography. The Rf and retention time correlated with known cold standard SKI230.
Tumor Cell Line Models
The human origin cell lines K562 (CML) and BT-474 (breast ductal carcinoma) were used for the assessment of Abl PKIs, based on the previously reported analysis of the expression of the Abl gene (25). A-431 (epidermoid carcinoma) cells were also used for comparison in Western blot analysis. The cells were obtained from the American Type Culture Collection and handled in tissue culture according to the vendor's instructions. The K562 cells were grown in suspension culture in RPMI supplemented with 10% fetal bovine serum (FBS) in a humidified atmosphere (e.g., 37°C, 5% CO2). The BT-474 and A-431 cells were grown as monolayers in modified Dulbecco's medium (prepared in-house) containing 4 mM l-glutamine, 1.5 g/L of sodium bicarbonate, and 10% FBS.
Saturation Binding Studies
Saturation binding studies were used to evaluate for possible nonspecific binding of 131I-SKI230 to the protein kinase. Briefly, 12 test solutions were prepared containing increasing amounts of the 131I-SKI230 (no carrier added) ranging from 0 to 100 nM and 106 of K562 or BT-474 cell suspension in a total volume of 500 μL of 25 mM N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (HEPES) with 2% bovine serum albumin (BSA), pH 7.6. For each concentration of 131I-SKI230, nonspecific binding was determined in the presence of 1,000 nM SKI230. The solutions were gently shaken at ambient temperature for 1 h; the cells were isolated by rapid filtration through a glass fiber filter (Whatman #32; Whatman), washed with 2 × 3 mL of ice-cold Tris-buffered saline (10 mM Tris, 0.9% NaCl, pH 7.4), and counted in a γ-counter. The amounts of specifically bound SKI230 were plotted against the concentration of SKI230 (x-axis) and the data fitted to a 1-site saturation curve using a least-squares-regression method (Origin; MicroCal) to determine the dissociation constant and maximum number of binding sites. A Scatchard transformation was also performed.
Displacement Binding Studies
To assess the avidity of 131I-SKI230 binding to the target Abl site in the living cells, 12 test solutions were prepared (in triplicate), each containing radiolabeled 131I-SKI230 in the amount equal to 20,000 counts per minute (cpm) and in increasing amounts of either unlabeled SKI230 or imatinib (0.001–1,000 nM) and 5 × 105 K562 and BT-474 cells in a total volume of 500 μL of 25 mM HEPES (2% BSA, pH 7.6). The incubation, harvesting, and counting were performed as for saturation binding studies. The amount of bound 131I-SKI230 was plotted against the concentration of SKI230 or imatinib (x-axis) and the data fitted to a sigmoidal function using a least-squares-regression method (Origin; MicroCal) to determine the inhibitory concentration of 50%.
Kinetic Binding Studies
Kinetic binding studies were performed to assess the time-dependent kinetic profile of SKI230 binding. Six test solutions were prepared (in triplicate) containing 20,000 cpm of 131I-SKI230 and 5 × 105 K562 cells in a total volume of 500 μL of 25 mM HEPES (2% BSA, pH 7.6). The solutions were gently shaken at ambient temperature. A second set of samples in a total volume of 450 μL was prepared and gently shaken at ambient temperature. At the end of 1 h, 50 μL of 10 μM SKI230 were added, and the tubes were shaken at ambient temperature. At various times, triplicate samples were removed and the cells isolated by centrifugation, washed with ice-cold phosphate-buffered saline, and counted in a γ-counter. The cell-bound 131I-SKI230 was plotted against the incubation time.
Western Blot
Western blot was performed using anti–c-Abl polyclonal rabbit antibodies (Sigma) in a 1:2,000 dilution and an ECL Western blotting analysis system (AmershamPharmacia Biotech).
Protein Binding Study
The fraction of the radiolabeled compound nonspecifically bound to proteins was determined by ultrafiltration (Centricon YM-10; Amicon Centrifugal Filter Devices) (10,000 molecular weight cut-off) of dilutions of FBS ranging from 1% to 10% containing 3.7 kBq/mL (0.1 μCi/mL) of 131I-SKI230. A control study was done in saline to ensure that the activity did not passively bind to the filter. The ultrafiltration devices were centrifuged at 1,500g for 30 min at 23°C to collect approximately 30% of the original volume of serum. The percentage of protein-bound activity was calculated.
In Vitro Drug Sensitivity Assessment
A WST-1 cell proliferation assay reagent (Roche Diagnostics) was used to assess the response of the cells to the PKI treatment. Cells (3,000/well) were plated in 96-well plates in a final volume of 100 μL/well culture medium and treated with SKI230 at a dose range from 1 pM to 1 μM continuously for 72 h. Then, 10 μL of WST-1 reagent was added to the wells. After 30–50 min of incubation, the colorimetric data were acquired at absorbance wavelength (λmax = 440 nm) against a background reference wavelength of 595 nm using a Spectra Count microtiter plate reader (Packard/Perkin-Elmer) (23). The effective concentration of 50% (EC50) was calculated using a nonlinear regression fit to a sigmoid dose-response curve using Prism 4 (GraphPad Software).
In Vitro Radiotracer Accumulation Assay
The radiotracer uptake assay was performed as previously described (24). The cells were exposed to medium containing 131I-SKI230 (3.7 kBq/mL [0.1 μCi/mL], no carrier-added), cells and medium samples were harvested, and cell pellets were weighed and radioactivity assayed by γ-spectrometry. The data were expressed in terms of cpm/g of cells and cpm/mL of medium, and the cell-to-medium ratios of weight-normalized counts were plotted over time to estimate the kinetics of 131I-SKI230 accumulation in cells.
SiRNA Abl Inhibition Confirmation
An Abl siRNA/siAb assay kit and siIMPORTER transfection reagents and protocols (Millipore) were used. The K562 cells were incubated in medium containing 100 or 200 nM siRNA in flasks or 6-well microplates and the transfection reagent for 72 h. Mixed nonspecific RNA provided with the kit (control pool) was used as a negative control. Then a radiotracer uptake study was performed with 131I-SKI230 over 1 h (as described). The results were normalized by the number of cells. Radiotracer accumulation ratios were compared for the Abl siRNA–transfected cells, wild-type cells, and negative control cells exposed to nonspecific siRNA control pool. The data were expressed as a percentage to the wild-type cell uptake values.
Animal Tumor Model
All animal studies were performed under an Institutional Animal Care and Use Committee–approved protocol (no. 86-02-020). Animals were under anesthesia during image aquisition using 2% (at 1 L/min) isoflurane (Forane; Baxter Healthcare). Subcutaneous tumor models in rnu/rnu rats (Frederick Cancer Institute) were used to assess the feasibility of 124I-SKI230 for in vivo PET of BCR-Abl expression. K562 cells were used for the generation of the ectopic xenograft tumors. The tumor cells from the cell culture were resuspended in the Matrigel (BD Biosciences) and injected subcutaneously in the shoulder as a 5 × 106 cells dose. Tumor growth was monitored daily, and the imaging sessions were scheduled on the smallest tumor (reaching the size of 1 cm3) in the animal.
In Vivo Radiotracer Administration
Cold NaI solution (0.5 mL/rat, intraperitoneally, 0.9% w/v) was injected 15 min before the administration of the radiotracer. Syringes with Teflon (DuPont) pistons were used to reduce 124I-SKI230 surface binding. 124I-SKI230 was administered intravenously at a dose of 7.4 MBq (200 μCi) per animal. Tissue sampling for the ex vivo assessment of radiotracer accumulation was performed after animal euthanasia at the end of 90 min of imaging.
Clinical PET
The animal, under general deep intravenous sedation (ketamine/acepromazine), was placed in the geometric center of the detector ring. Images were acquired using a dynamic acquisition protocol with 10-min frames over 90 min of observation, using the Advance tomograph (GE Healthcare), with a spatial resolution of 5 mm in full width at half maximum (FWHM) at the center of the field of view. This camera has previously been cross-calibrated with the AutoGamma 5550 spectrometer (Packard). Reconstruction was performed using the filtered backprojection algorithm. Attenuation correction was performed using transmission images.
Emission counts were corrected for random coincidences, dead time, and scatter. Emission scans were reconstructed using an iterative method, with measured attenuation correction and smoothed with an 8-mm gaussian filter. The reconstruction parameters were 28 subsets and 2 iterations in a 256 × 256 matrix using a loop filter of 2.15 mm in FWHM and a postprocessing filter of 3.0 mm in FWHM. Regional tumor radioactivity concentrations (percentage injected dose per gram [%ID/g]) were estimated from the maximum pixel within regions of interest (ROIs) drawn around the tumor on transaxial slices of the reconstructed image sets. Maximal standardized uptake values were measured using software (GE Healthcare) with 3-dimensional ROIs drawn on orthogonal reconstruction. The signal-to-background ratio was calculated to assess the specificity of the tracer accumulation.
In Vivo Uptake Blocking Studies
K562 or A-431 cells (3 × 107) mixed with Matrigel (BD Biosciences) were inoculated into the right shoulder region of 3 athymic rnu/rnu rats (National Cancer Institute).
Three weeks after tumor inoculation, in vivo uptake blocking studies were performed using a Focus microPET scanner (Concorde Microsystems), in 2 stages. During the first stage, baseline SKI230 in vivo binding to the CML tumor model was assessed. Three to five seconds after the start of dynamic small-animal PET, 18.5–22.2 MBq of 124I-SKI230 were injected intravenously in 200 μL. The dynamic image was acquired over 120 min at 250–750 keV and reconstructed using filtered backprojection without attenuation correction.
Three days later, the test image was acquired before the second stage to ensure that no significant activity remained within the animal and in the tumor. At the second stage, the same animals were injected with 1,000 nM of unlabeled SKI230 at 5 min before the repeated administration of 124I-SKI230 and dynamic image acquisition. The viability of the xenograft was confirmed on all animals using a 5-min small-animal PET image acquisition with 18F-FDG (Eastern Isotopes) 2 d after the displacement studies (data not shown). The visualization and analyses of small-animal PET images, including the calculation of %ID/g measurements (26), were performed at 120 min using AsiPRO software (Concorde Microsystems), with values adjusted according to in-house phantom studies.
Statistical Analysis
For paired comparisons of samples, the χ2 and Fisher exact tests were applied. Error bars on plots reflect 95% confidence interval.
RESULTS
Radiochemical Purity
131I-SKI230 and 124I-SKI230 were synthesized with a specific activity of 37 kBq/nM (1 μCi/nM) and purified to greater than 99% for each study.
Nonspecific Protein Binding Affinity of SKI230
In vitro cell-free binding assessment demonstrated high binding of the 131I-SKI230 (1 μCi/nM) to the proteins in the ultrafiltrated sera. At 1% FBS concentration, the protein-bound fraction of the tracer increased to 33.5%, and at 10% FBS the protein-bound tracer fraction reached 53%.
Competition Binding Study
To confirm that the radiolabeled compound had a high specific affinity to BCR-Abl, competitive binding studies were performed in K562 cells. The studies showed rapid saturable accumulation of 131I-SKI230 in the tumor cells overexpressing Abl (Fig. 1B). A washout of a fraction of the initially accumulated radiotracer after the addition of the cold SKI230 at micromolar concentration (dotted plot) is consistent with binding specificity. It also indicates a rapid equilibrium underlying the accumulation of SKI230 in the cells with rapid off (4.6%/min) and on (4.4%/min) rates (Fig. 1B).
Saturation Binding Studies
Both a saturation curve and a Scatchard plot for the SKI230 binding are shown in Figure 1C. A total of 42,180 ± 1,950 (P = 0.05) binding sites per cell were found. The binding was of high affinity, with a dissociation constant of 9.8 ± 0.6 nM (P = 0.05).
Displacement Studies
Displacement studies compared cold SKI230 and imatinib and indicate that SKI230 has 6.1 ± 0.7 (P = 0.05) higher affinity (Fig. 1D) to BCR-Abl than does imatinib. Imatinib displaces SKI230, probably from a single high-affinity site (Fig. 1D).
In Vitro Radiotracer Accumulation Studies
To study the relative uptake of radiotracer in BCR-Abl–expressing versus control cells, an uptake study was performed with K562 and BT-474 cells growing in tissue culture. Significantly higher cell-to-medium ratios of 131I-SKI230 accumulation were observed in cultured living K562 cells that express high levels of BCR-Abl than in BT-474 cells in which BCR-Abl was not detectable by Western blot techniques (Fig. 2A). Both cells had some degree of uptake, with the concentration in K562 cells plateauing at a cell-to-medium ratio of 120 mL/g and BT-474 at a cell-to-medium ratio of around 40 mL/g. Presumably the uptake in the control was due to nonspecific binding secondary to SKI230 lipophilicity. A plateau of accumulation was observed after 60 min of incubation with the tracer. Low-rate accumulation was observed between 10 and 60 min. The cell uptake of 131I-SKI230 at equilibrium (over 1 h) was 2- to 3-fold higher in K562 cells than in BT-474 cells.

In vitro characterization of PKI for direct imaging of Abl expression in tumors. (A) Radiotracer accumulation assay. In vitro uptake studies in cell lines K562 (overexpressing BCR-Abl) and BT-474 (devoid of Abl expression). (B) Inhibitory concentrations of 50% for SKI230 in cytotoxicity assay (WST-1 colorimetric assay). (C) Proof of SKI230 binding specificity using Anti-Abl siRNA in radiotracer accumulation assay: wild-type K562, (N1); K562 transfected with nonspecific siRNA control pool (N2); K562 transfected with 100 nM anti-Abl SiRNA (N3); and K562 transfected with 200 nM anti-Abl SiRNA (N4). (D) Western blot analysis of cells treated with radiolabeled compound. 1, Abl antibody Western blot anti–Human-c-Abl; 2, protein standards; 3, A-431; 4, BT-474; 5 and 6, K562 (6, control for siRNA experiment); 7, K562 after 100 nM siRNA; and 8, K562 after 200nM siRNA.
In Vitro Assessment of Abl Kinase Inhibitor Cytotoxicity
In vitro assessment of the drug activity of the cold compounds demonstrated a correlation between the in vitro radiotracer uptake and the ability of the cold SKI230 compound to inhibit cell proliferation. Figure 2B shows that the K562 cell line overexpressing BCR-Abl with a high level of radiotracer uptake (Fig. 2A) was highly sensitive to SKI230 (EC50, 8.5–9 nM), whereas the control breast cancer cell line BT-474 was much less sensitive to the drug (EC50, 2.4 μM).
Proof of SKI230 Specificity Using Anti-Abl siRNA
To prove the specificity of SKI230 uptake in the cells related to BCR-Abl expression, we studied the effect of siRNA directed against the Abl kinase domain on the degree of the radiotracer accumulation within the tumor cells (Fig. 2C). The studies of SKI230 specificity performed using siRNA silencing Abl expression demonstrated a 3-fold drop of the PKI accumulation in the K562 cells with siRNA-treated cells (Fig. 2C), as measured by the 131I-SKI230 uptake. On the basis of both Western blot assessment of Abl kinase expression (Fig. 2D) and a corresponding effect on the radiotracer binding (Fig. 2C), the siRNA had a dose-dependent effect on Abl kinase expression.
Western Blot for Abl Expression
Figure 2D confirmed the high level of Abl expression in the CML cell line K562 and low level of expression in BT-474 and A-431 cell lines used as negative controls.
In Vivo Imaging of Radiotracer Kinetics
The kinetics of in vivo radiotracer distribution were assessed in a series of in vivo imaging experiments in rats bearing Abl kinase–expressing (K562) human tumor xenografts on the shoulder (Fig. 3). Rapid accumulation of 124I-SKI230 was observed in the K562 tumor. Comparison of the images acquired at different time points demonstrated a gradual increase in the radiotracer accumulation in tumor xenografts over a 90-min period, maximizing at the end of the observation period (Figs. 3A and 3B). The maximum signal-to-background ratio for the specific accumulation in the tumors was also registered at the 90-min time point (Figs. 3A–3C). ROI analysis demonstrated progressive accumulation of activity in the tumor, and clearance of the arterial input signal was observed in the projection of the heart (Fig. 3C). Interestingly, the uptake profile in the background muscle tissue remained flat, which—in combination with progressive accumulation of radiotracer in the tumor—resulted in the improvement of tumor-to-background ratio over the time of observation.

Direct in vivo imaging of Abl expression in rat xenograft human CML model. (A) Time series of axial PET Advance images of rat, bearing tumor model K562 overexpressing BCR-Abl representing biodistribution of 124I-SKI230 in animal. (B) Sagittal projection of accumulation of 124I-SKI230 in Abl-expressing CML tumor model in shoulder area 90 min after administration of direct PKI imaging agent. (C) Pharmacokinetic profile of 124I-SKI230 biodistribution in organs and tissues quantified using image analysis of radiotracer accumulation based on ROI analysis of dynamic frames. (D) Ex vivo well counts calculated for tissue samples obtained by postmortem tissue collection (at 90 min) confirming SKI230 pharmacokinetic results.
The hepatobiliary clearance dominated for 124I-SKI230 in the rat model. Radiotracer accumulated in the liver early, peaked at 10 min, and then gradually cleared, with a corresponding increase in the small bowel–excreted activity (Figs. 3A and 3C). Renal clearance, visualized as reduction of kidney signal with growth of bladder activity, comprised less than 30% of radiotracer excretion (Figs. 3A and 3C).
Ex vivo tissue sampling (Fig. 3D) confirmed that the clearance of 124I-SKI230 occurred primarily through the hepatobiliary system into the intestine, exceeding the level of 5% of the dose per gram in the liver at 90 min. Tumor uptake by 90 min increased to 1.3%/g. At that time point, the tissue-to-background ratio calculated relative to muscle was 4.1.
Direct In Vivo Imaging of Abl Protein Kinase Inhibitor Treatment of CML: Blocking of 124I-SKI230 In Vivo Uptake Within Engrafted Tumor
Figures 4A and 4B show transaxial images of a representative rat (n = 3). The images were acquired before and after systemic treatment with SKI230. 124I-SKI230 uptake in A-431 did not differ significantly from uptake in muscle (Fig. 4D). Comparison of in vivo accumulation of 124I-SKI230 in the animal model of CML before (Fig. 4A) and after (Fig. 4B) a single treatment with cold SKI230 demonstrated a significant decrease in the intensity of radiotracer uptake (Fig. 4B) from a mean of 1.1 %ID/g to a mean of 0.5 %ID/g (Fig. 4C). The reduction in tracer activity within the tumor as a result of PKI treatment was about 54%. The reduction of uptake was not related to tumor death or apoptosis from a single dose of SKI230 treatment, as confirmed by 18F-FDG uptake (data not shown).

Blocking of 124I-SKI230 in vivo uptake within engrafted tumor. (A) Transaxial images of representative rat (n = 3) before PKI treatment. (B) Transaxial images of same rat after cold SKI230 administration, demonstrating in vivo displacement of radiotracer by micromolar concentration of systemically administered PKI. (C) 124I-SKI230 tumor uptake after blocking with cold SKI230 (open bars) is reduced, compared with pretreatment uptake (solid bars). (D) Imaging of negative-control A-431 tumor model.
DISCUSSION
SKI230 was designed in silica (24,27) specific for the ATP binding pocket of Abl protein kinase, which was initially demonstrated in K562 cells, overexpressing the BCR-Abl (24). The key role of BCR-Abl in the malignant transformation of CML cells and proven treatment success make it a potential target for nuclear imaging. Several other tumors were reported to express c-Abl: chondrosarcoma, liposarcoma, gastric adenocarcinoma (12,13), ovarian carcinoma (14), and oral squamous carcinoma (15). Some of them appeared responsive to treatment with Abl PKIs (6,7,16), suggesting the potential research and clinical role of the radiolabeled PKI imaging compound in the monitoring of treatment of CML and other types of cancer dependant on Abl kinase activity.
11C labeling of imatinib was patented by Collins, Klecker, and Anderson (28) for the imaging of the drug accumulation as a guide to antitumor therapy. In vivo pharmacokinetic imaging studies demonstrated the potential application of radiolabeled compound for the assessment of PKI biodistribution only (no tumor model) (20) in primates, with slow peaking uptake in the liver and kidneys, followed by clearance. In an attempt to develop radiolabeled Abl-specific PKI compounds, 11C-AG957 analogs were reported to reduce the proliferation of the FDrv210C cell line in vitro (29); however, uptake of the radiolabeled compound in tumor cells was not shown. In this study, we demonstrate the entire spectrum of in vitro assessment of the pharmacodynamic properties of the radiotracer, in vivo pharmacokinetics, and a model of potential clinical application in an in vivo displacement study.
In the preliminary studies of SKI230 (24), both in vitro cytotoxicity and accumulation were demonstrated in K562 and A-431 cells. Our quantitative comparison to imatinib showed a log higher binding affinity of SKI230, proving sensitivity of the radiolabeled SKI230 for imaging applications in tracer concentrations.
PET with radiolabeled dasatinib analog, described by Veach et al. (21), also demonstrated PKI biodistribution to the BCR-Abl–expressing tumor in vivo. The overall pattern of tumor targeting is similar to that in the current study. However, dasatinib imaging may play a rather important role in identification of the cases resistant to the first-line anti-Abl PKI therapy, whereas SKI230 can be used for the general screening purposes.
Further assessment of target specificity showed that, like most other pyridopyrimidinones, SKI230 demonstrated specificity to c-src (30). The homology of Abl and Scr tyrosine kinase domains was described earlier (31). Dual specificity of the compound should not interfere with its antitumor action, because Abl was shown to participate in the induction of c-src signal transduction pathway (30,32). We have tested the specificity of the anticancer action of SKI230 by comparing cell lines with known levels of Abl and c-src expression (25). The importance of the dual specificity of the pyridopyrimidinone compounds was emphasized in studies demonstrating improved control of the downstream S tat 5 and Erk cell survival and independent effect of PKI action on Abl and c-src pathways in K562 cells (30). Dual specificity of SKI230, however, may have some limitations for quantitation of PKI action in biologic systems with an unknown combination of Abl and c-src.
The current study confirmed the important role of Abl kinase inhibition in controlling cancer cell proliferation. Abl inhibitor treatment in vitro led to death of the tumor cell lines (32). In our experiment, tumor growth inhibition and displacement experiments proved a quantitative relationship between the 131I-SKI230 uptake and the cytotoxic effect of the treatment, with the cold compound providing background for quantitative PET assessment of PKI activity. The measurement of the number of binding sites may allow for the assessment of the therapeutic targets on the cancer cells and the potential toxicity to the healthy tissues.
The specificity of Abl kinase pathway involvement was confirmed through termination of synthesis of the specific tyrosine kinase by siRNA. This method currently is broadly used for the identification of specific signal transduction pathways (33–35). Abl-specific siRNA was shown to be efficient in vitro in the K562 cell line, reducing the number of BCR-Abl mRNA transcripts and lowering imatinib mesylate inhibitory concentration of 50% (36). SiRNA was also used as a proof of specificity in imatinib studies in microarray for downstream genes showing similarities in action to siRNA and PKI (37).
The use of radiolabeled PKIs as a direct imaging agent attempted with reversible EGFR inhibitors in vivo (18) was suboptimal because of a rapid tracer washout of tumor tissue. 11C-labeled irreversible EGFR inhibitors provided better imaging, but because of the short half-life of the radioisotope these inhibitors had time limitations for achieving an optimal signal-to-background ratio (19). Pal et al. (38) were successful in demonstrating 124I-IPQA for imaging EGFR tyrosine kinase signal transduction pathway in vivo. Using 124I for SKI230 labeling, we considered extending the radiotracer physical half-life to improve quality with delayed images: further specific accumulation in the tumor and clearance from the background tissues. 11C-labeled erlotinib was also reported to successfully image EGFR-expressing lung tumor models in mice (39).
In SKI230, we observed preferential distribution to the liver, as in other PKI imaging studies (20,21,40), because of high LogP values or lipophilicity resulting in the predominance of early liver uptake and hepatobiliary clearance. Lipophilicity also leads to nonspecific tissue distribution or binding (40). A more favorable pattern of biodistribution is observed in the radiolabeled clinically proven PKIs, such as imatinib (20) or dasatinib (21). However, some unexpected results may be observed, as seen in 18F-gefitinib—which had not only a rapid and predominantly hepatobiliary clearance but also a high nonspecific, nonsaturable cellular uptake, resulting in poor correlation with EGFR expression levels (40). Other potential ways of resolving bioavailability problems include PEGylation (41) and lipophilic microspheres.
Future Applications in Drug Screening
With a great number of emerging potential therapeutic compounds, an effective screening method is critical to increase cost-effectiveness and reduce bench-to-bed time. Recently, a solid-phase PKI screening method in multiwell plates was suggested using an imatinib analog (42). Displacement studies reported in the article demonstrate one of the potential routes to the in vitro application of the radiolabeled SKI230.
Future Applications in Patient Selection
High specificity of SKI230 to BCR-Abl may facilitate noninvasive assessment of potential candidates for PKI treatment, pending proof of tracer affinity correlation with treatment effect or overall patient prognosis.
Future Applications in Treatment Assessment
The 18F-FDG PET approach proved effective in the indirect imaging of PKI treatment response in gastrointestinal stromal tumors (43) and CML (44). C-Kit–mediated AKT-dependent regulation of glucose uptake by glucose transporter 3 and glucose transporter 4 and hexokinase activity was proven to be a specific mechanism responsible for both clinical and imaging effect of treatment (45,46) in gastrointestinal stromal tumors. Radiolabeled Abl PKI (124I-SKI230) provides a pathway-specific tool to show drug–target interaction. Minimizing the potential interference from the intermediate steps could lead to better specificity of PET treatment response assessment (47).
CONCLUSION
Concentration-dependent displacement of the radiolabeled compound was demonstrated in the competition, potentially allowing for quantitation of the drug concentration in tissues, if the radiotracer is administered during a therapeutic PKI treatment. The labeling of SKI230 with an isotope of longer half-life, such as 124I, was beneficial, permitting 2 imaging sessions with potential quantification of the treatment effect.
In the future, noninvasive molecular imaging with PET may accelerate clinical development of kinase inhibitors.
Acknowledgments
We acknowledge the contribution of Ronald Finn, for assistance in radiosynthesis, and Mohammad Namavari, for assistance in radiolabeling. The study was supported by the following grants: P50 CA086438-05S2 from ICMIC and R01 DOEN86ER-60407-9 and P01 CA094060-05 from DOE. Technical services provided by the MSKCC Small-Animal Imaging Core Facility, supported in part by grant R24 CA083084-09 from NIH Small-Animal Imaging Research Program (SAIRP) and grant P30 CA08748 from NIH Center, are gratefully acknowledged.
Footnotes
-
COPYRIGHT © 2010 by the Society of Nuclear Medicine, Inc.
-
↵* Contributed equally to this work.
References
- Received for publication October 12, 2007.
- Accepted for publication September 8, 2009.





{kind=link}
{kind=link}
{kind=link}
{kind=link}