


Abstract
The purpose of the present study was to measure adenosine A2A receptor (A2AR) occupancy in the brain by a novel adenosine A1/A2A antagonist, 5-[5-amino-3-(4fluorophenyl)pyrazin-2-yl]-1-isopropylpyridine-2(1H)-one (ASP5854), and to determine the degree of receptor occupancy necessary to inhibit haloperidol-induced catalepsy in rhesus monkeys. Methods: A2AR occupancy by ASP5854 (0.001–0.1 mg/kg) was examined in the striatum using an A2AR-specific radiotracer, 11C-SCH442416, and PET in conscious rhesus monkeys. A2AR occupancy was monitored after a single intravenous administration of ASP5854 in 3 animals, and a dynamic PET scan was performed at 1, 4, and 8 h after an intravenous bolus injection of the tracer for approximately 740 MBq. Catalepsy was induced by haloperidol (0.03 mg/kg, intramuscularly) and examined for incidence and duration. Results: ASP5854 dose-dependently increased A2AR occupancy in the striatum and showed long-lasting occupancy even after the reduction of plasma concentration. Haloperidol induced severe catalepsy at 40 min after intramuscular injection. The incidence and duration of cataleptic posture were dose-dependently reduced by ASP5854 at 1 h after oral administration, and the minimum ED50 value was 0.1 mg/kg. Administration of a dose of 0.1 mg/kg yielded a plasma concentration of 97 ± 16.3 ng/mL, which corresponded to 85%–90% of A2AR occupancy. Conclusion: These results showed that ASP5854 antagonized A2AR in the striatum, and the dissociation from A2AR was relatively slow. In addition, more than 85% A2AR occupancy by ASP5854 resulted in an inhibition of haloperidol-induced catalepsy. Thus, such a pharmacodynamic study directly demonstrates both the kinetics of a drug in the brain and the relationship between dose-dependent receptor occupancy and plasma level.
Adenosine A2A receptors (A2ARs) are abundantly localized in the caudate-putamen, nucleus accumbens, and olfactory tubercle in several species (1). They are coexpressed with dopamine D2 receptors in the GABAergic striatopallidal neurons; in contrast, there are no A2ARs in the neurons projecting from the striatum to the substantial nigra and expressing D1 receptors (2,3). Stimulation of adenosine A2ARs decreases the binding affinity of D2 receptors (4) and elicits effects opposite to the ones shown by D2 receptor activation at the level of second-messenger systems and early gene expression (5,6). These observations suggest that antagonistic adenosine–dopamine interactions may be important in the regulation of the activity of the basal ganglia and could explain the depressant and stimulating effects of adenosine A2AR agonists and antagonists on motor behavior (7).
In addition, adenosine receptor agonists induce sedation and catalepsy dose-dependently and inhibit the motor-activating effects of dopamine receptor agonists (7,8). In contrast, adenosine receptor antagonists, including caffeine and related methylxanthines, produce motor-stimulant effects (9), which appear to be related to an action on A2A rather than on A1 receptors (10). More recently, the A2A selective antagonist KW-6002 has been documented to remit adenosine A2AR agonist-, haloperidol-, and reserpine-induced catalepsy in rodents as well as motor impairment in nonhuman primate models of Parkinson's disease (PD) (11,12). These findings support a role of A2ARs as neuromodulators of dopaminergic function and suggest that they play an important role in movement disorders such as PD.
We have recently identified 5-[5-amino-3-(4fluorophenyl)pyrazin-2-yl]-1-isopropylpyridine-2(1H)-one (ASP5854) as a novel and potent, centrally active adenosine A1 and A2A dual antagonist from our optimization screening of pyrazolopyrimidin derivatives (13). ASP5854 inhibited haloperidol-induced catalepsy in rodents, induced an increase in the contralateral turning behavior caused by a subthreshold dose of l-3,4-dihydroxyphenylalanine in rats with a unilateral 6-hydroxydopamine lesion of the dopaminergic nigrostriatal pathway (13), and improved motor impairment in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine–treated marmosets (Takuma Mihara, unpublished data, 2008). These effects of ASP5854 include the antagonism for adenosine A2ARs.
PET enables the in vivo study of several physiologic and neurochemical variables using methods originally developed for quantitative autoradiography (14). Results from the imaging data using nonhuman primates together with plasma concentration of drug can be used to examine the plasma level–receptor occupancy relationship and can be useful for the determination of dosing regimens in human trials. Several adenosine A2A antagonists have been used as labeling ligands with positron-emitting radioisotopes (15–17). Among these antagonists, xanthine derivatives had trouble with processes such as nonspecific binding and photoisomerization (18,19), whereas nonxanthine derivatives avoided these problems. 11C-SCH442416 has been recently introduced as the first, to our knowledge, nonxanthine ligand suitable for in vivo imaging of adenosine A2AR in PET because of the high affinity and selectivity to adenosine A2AR of the compound, the good signal-to-noise ratios, and a low amount of radioactive metabolite in the brain of nonhuman primates (20). Thus, this compound might be useful for real-time imaging of receptor occupancy via PET in a living-animal experimental study.
The purpose of the present study was to quantify the adenosine A2AR occupancy in nonhuman primates after the administration of various doses of ASP5854 using 11C-SCH442416 via PET. In addition, the study was performed to demonstrate the degree of receptor occupancy required to achieve consistent haloperidol-induced catalepsy in the pharmacokinetic–pharmacodynamic relationship.
MATERIALS AND METHODS
Materials
ASP5854 was synthesized by Fujisawa Pharmaceutical Co., Ltd. (now Astellas Pharma Inc.). An adenosine A2A–specific radiotracer, 11C-SCH442416, was synthesized using an automated apparatus (17). Five milligrams of haloperidol (Serenace) were purchased from Dainippon Sumitomo Pharma Co., Ltd., and heparin sodium injection-N (1,000 units/mL) was purchased from Ajinomoto Pharma Co., Ltd.
Measurement of Adenosine A2A Receptor Occupancy Using PET in Rhesus Monkeys
The monkeys were maintained and handled in accordance with the National Institutes of Health recommendations, and all animal experiments were performed in compliance with the regulations of the Animal Ethics Committee of the Medical and Pharmacologic Research Center Foundation and Astellas Pharma Inc.
Three male rhesus monkeys were used for PET study (weight, 5.47 ± 1.84 kg; age, 5.0 ± 1.7 y). PET scans were performed with a high-resolution animal PET scanner (SHR-7700; Hamamatsu Photonics K.K.), with a transaxial resolution of 2.6 mm in full width at half maximum in the center of the scan field and a center-to-center distance of 3.6 mm (21). A 68Ge–68Ga blank scan (120 min) was performed before each study. During catheterization of the femoral vein for tracer injection and drug administration, each animal was transiently anesthetized with about 2% sevoflurane in a N2O/O2 gas mixture (7:3 ratio of N2O to O2). After the catheterization, anesthesia was immediately discontinued. Each monkey's head was fixed to a chair for PET scans with a head holder and stereotactically aligned parallel to the orbitomeatal plane with a laser marker; then a 68Ge–68Ga transmission scan (30 min) was performed. In the dose-dependent study, ASP5854 (0.01, 0.1, or 1 mg/kg) or saline (baseline condition) was intravenously administered after the transmission scan. One hour after the drug or saline administration, 11C-SCH442416 (∼740 MBq) was intravenously injected and a dynamic PET emission scan (10 s × 18 frames; 1 min × 7 frames; 5 min × 2 frames; total, 20 min, 27 frames) was simultaneously initiated. In the interaction between plasma concentration and occupancy for the adenosine A2AR study, 11C-SCH442416 (∼740 MBq) was intravenously injected and a dynamic PET emission scan was simultaneously initiated at 4 and 8 h after the drug administration. Although the animals' ears were unplugged during the emission scan, their eyes remained open under a dimmed light. In both experiments, the plasma sample for each PET scan was collected to measure the plasma levels of ASP5854.
Dynamic PET data were reconstructed with a Hanning convolution filter of 4.5 mm in full width at half maximum. Images were anatomically standardized using NEUROSTAT (22–24). From the dynamic images, binding potential (BP) was estimated for the striatum using a nonlinear least-square fitting procedure (the Marquardt algorithm (25) using a 3-parameter [R1, k2r, BP] simplified reference-tissue model (26) with the cerebellum as a reference region). Adenosine A2AR occupancy was calculated as a falling rate of the BP in a medicated condition from baseline in each animal.
Haloperidol-Induced Catalepsy in Rhesus Monkeys
Four adult male rhesus monkeys (weight, 4.79 ± 0.24 kg; age, 5.9 ± 0.1 y), purchased from Hamri Co. Ltd., were used in the study. The animals were tested 1 d per week, separating each testing day to allow for drug washout. On testing days, the animals were tested one at a time. The monkeys were brought into a quiet room and secured loosely in a primate chair (the primate chair had 4 clamps to fix 2 horizontal wooden bars [2.5 cm in diameter]; the position of the bars [the height, angle, and so on] was regulated so the animals could grip a bar with both hands and feet). Approximately 30 min after habituation on the experimental room, ASP5854 or 0.5% methylcellulose solution (2 mL/kg) was administered to the animal through an oral catheter (Izumo Health Co., Ltd.). After 20 min, haloperidol (0.03 mg/0.1 mL/kg) was given to the animal by intramuscular injection. All experiments were video-recorded to measure catalepsy. At 40 min after haloperidol injection, the animal's hands and feet were placed on and gripped the horizontal wooden bar. All animals were given 2 trials at 1-min intervals. This forced posture was not natural for the normal monkey, and if the monkey maintained the cataleptic posture, the trial was scored “onset-positive” for induced catalepsy. Otherwise, the trial was scored “onset-negative.” For onset-positive, the duration of maintaining cataleptic posture (defined as immobility, with eyes open, usually accompanied by unusual postures including rigid limb extensions or a twisted torso) was recorded as measures of the intensity of catalepsy during 5 min, determined by using video-recorded results after testing. A within-subjects crossover design was used.
Two months after the final catalepsy test, the same 4 animals were used to measure the concentration of ASP5854 in plasma. The animal was seated in the primate chair and habituated for 2 h. After the habituation, a 0.1 mg/kg dose of ASP5854 suspended in 0.5% methylcellulose was administered orally. The plasma sample was collected at 1 h after dosing from the upper limb vein, and heparin was added to the sample (final concentration, 2–5 units/mL) to inhibit coagulation.
Measurement of Plasma Concentration of ASP5854
Plasma concentration of ASP5854 was determined by a liquid chromatography–tandem mass spectrometric (LC-MS/MS) method. Deuterium-labeled ASP5854 was used as an internal standard. A 0.2-mL aliquot of plasma sample was mixed with 0.1 mL of a 5 ng/mL internal standard solution and 0.05 mL of water. This mixture was then applied onto a 30-mg/1-mL solid-phase extraction cartridge (OASIS HLB; Waters). After washing with 1 mL of water and 1 mL of water–methanol solution (6:4, v/v), the cartridge was eluted with 1 mL of methanol. The eluate was evaporated to dryness under a stream of nitrogen gas, and the residue was reconstituted in 0.2 mL of a mobile phase consisting of methanol, acetonitrile, water, and formic acid (700:300:1,000:1, v/v/v/v). A 10-μL aliquot of the resulting solution was then injected into an LC-MS/MS system equipped with a high-performance liquid chromatography system (Nanospace SI-2; Shiseido) and a mass spectrometer (TSQ Quantum Ultra; Thermo Fisher Scientific). High-performance liquid chromatographic separation was achieved using a 2.1-mm ID × 100 mm 5-μm column (Hypersil Gold; Thermo Fisher Scientific) that was eluted with the mobile phase described above at a flow rate of 0.2 mL/min. Mass spectral analysis was performed in positive ion electrospray ionization mode and with multiple-reaction monitoring (mass ranges, m/z 325 to 283 for ASP5854 and m/z 329 to 287 for the internal standard).
Statistical Analysis
In the haloperidol-induced catalepsy test, statistical analysis between the vehicle-treated group and the drug-treated groups was performed using the Steel test. ED50 value was calculated by the linear regression method.
RESULTS
Occupancy for Adenosine A2A Receptors by ASP5854
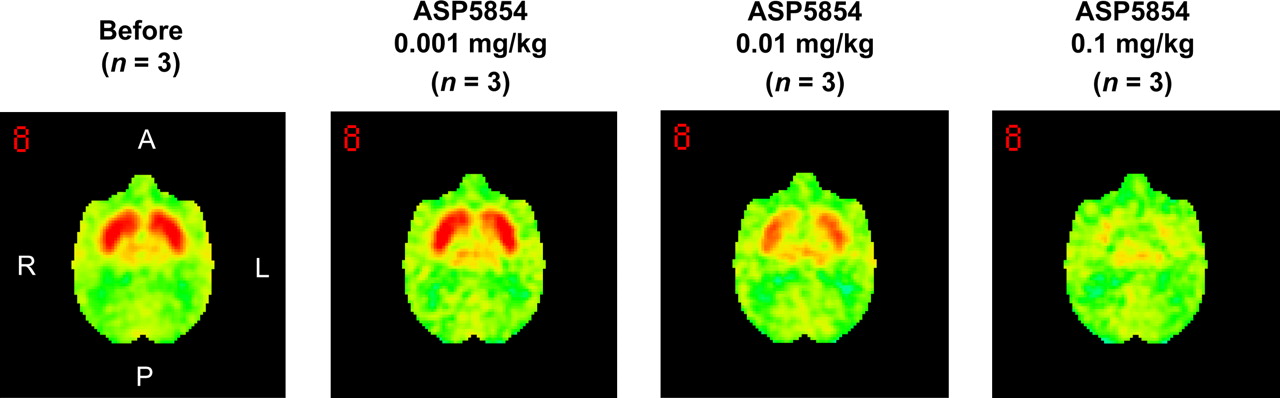
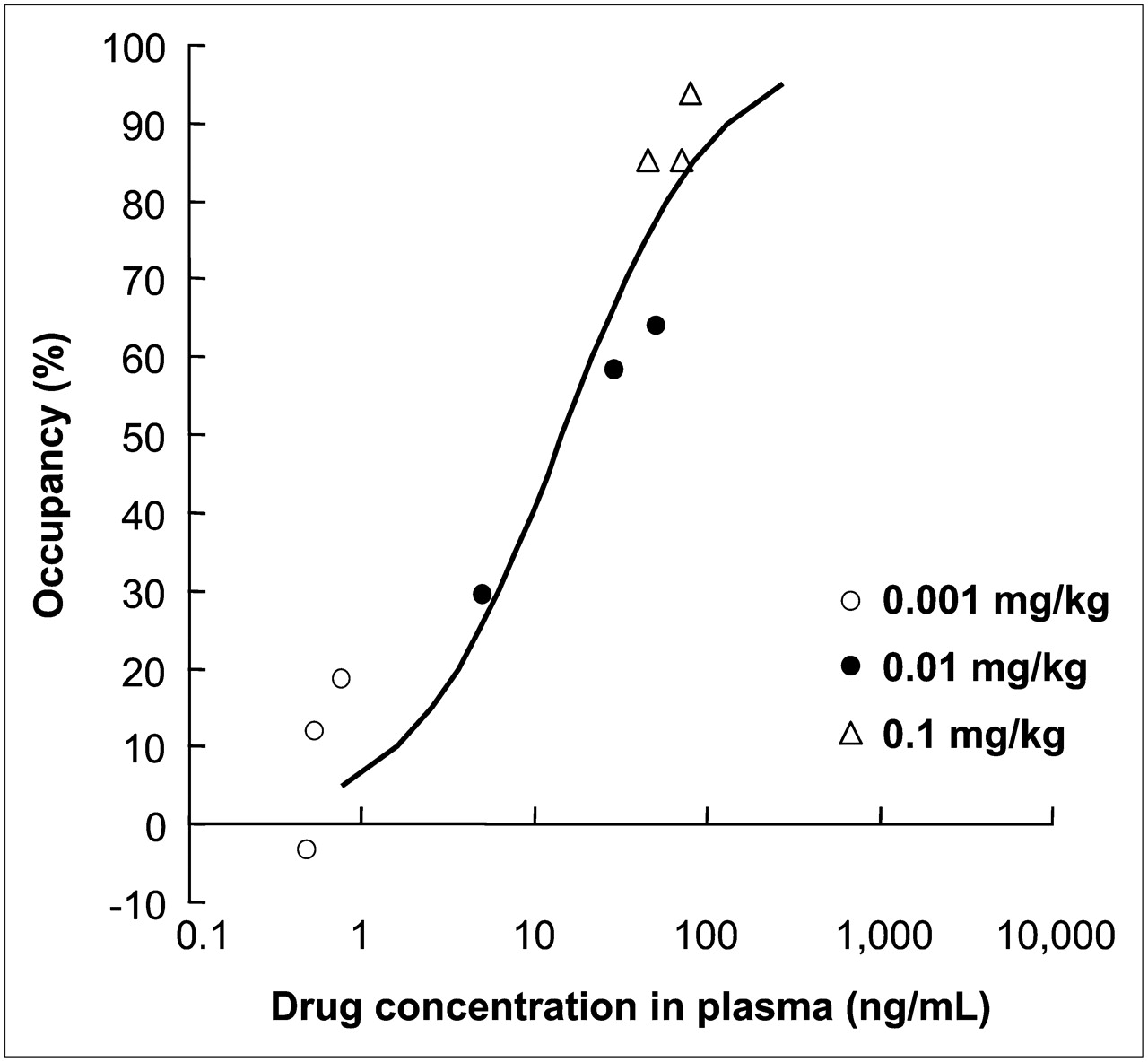
The mean and SD value of BP in the striatum at the baseline scan for each animal was 0.52 ± 0.06. The elective adenosine A2AR antagonist 11C-SCH442416 showed a high level of specific binding in the striatum, the area that reportedly has the highest expression level of adenosine A2AR in the brain; the averaged brain images from 3 animals are shown in Figure 1. This specific binding was displaced by an intravenous injection of ASP5854 in a dose-dependent manner (Fig. 1). At 1 h after the administration, the mean plasma concentration of each dose of ASP5854 (0.001, 0.01, and 0.1 mg/kg, intravenously) was 0.6, 28.6, and 64.4 ng/mL, and the occupancy for adenosine A2AR in the caudate was 9.0%, 50.7%, and 87.4%. A fitting curve of the plasma concentration compared with striatal receptor occupancy is shown in Figure 2. At 8 h after the administration, the plasma concentration of 0.1 mg/kg was reduced by approximately half compared with its level at 1 h after the administration; however, the receptor occupancy maintained a mostly even level up to 8 h after the administration (Fig. 3).

Averaged PET images of 11C-SCH442416 from 3 animals, from 5 to 10 min after tracer administration. Saline or ASP5854 (0.001–0.1 mg/kg) was intravenously administered at 1 h before 11C-SCH442416 administration. Images were anatomically standardized using NEUROSTAT and presented in cross-sectional images at striatum level.
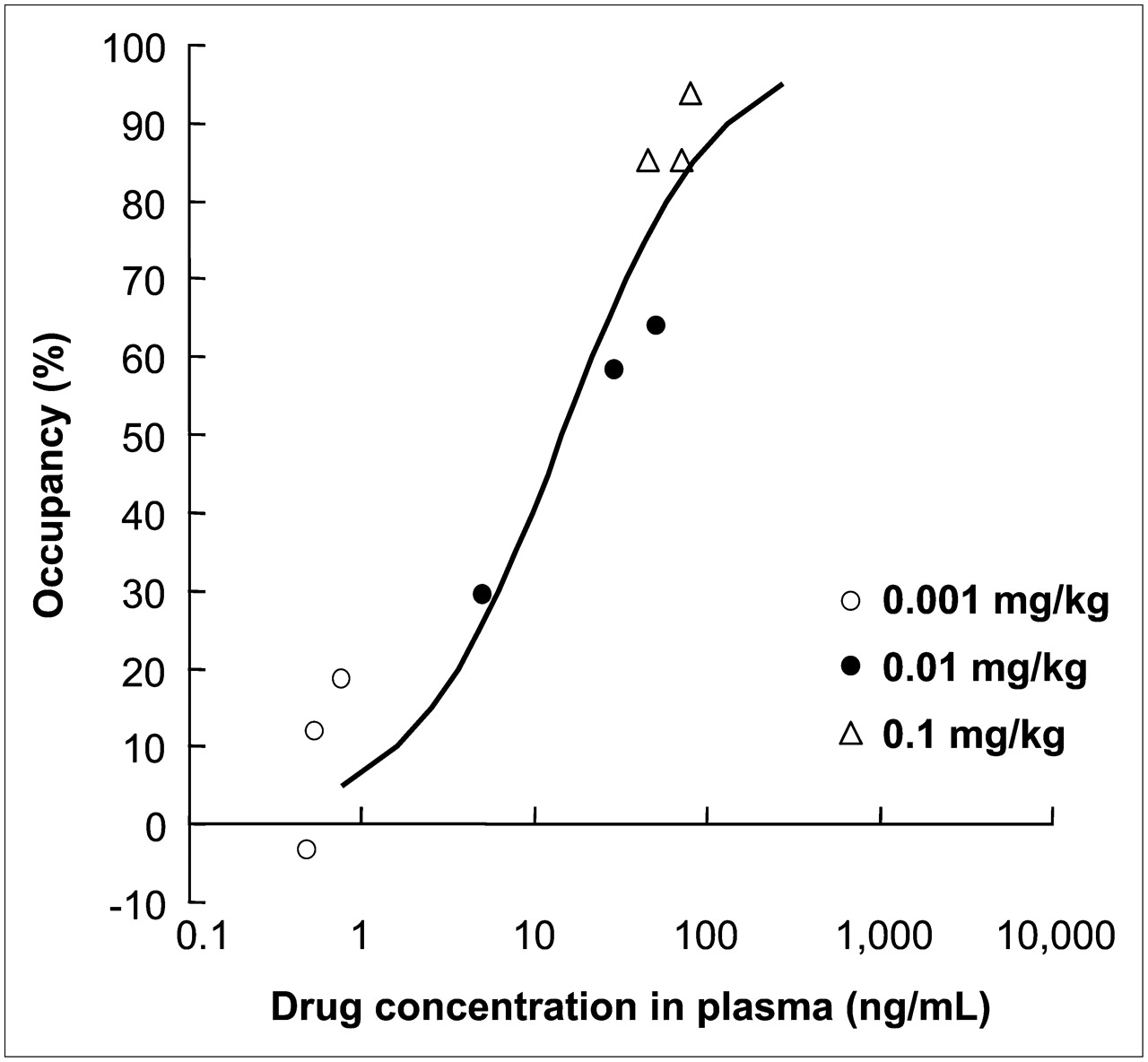
Fitting curve of plasma concentration and occupancy for adenosine A2AR in rhesus monkeys.
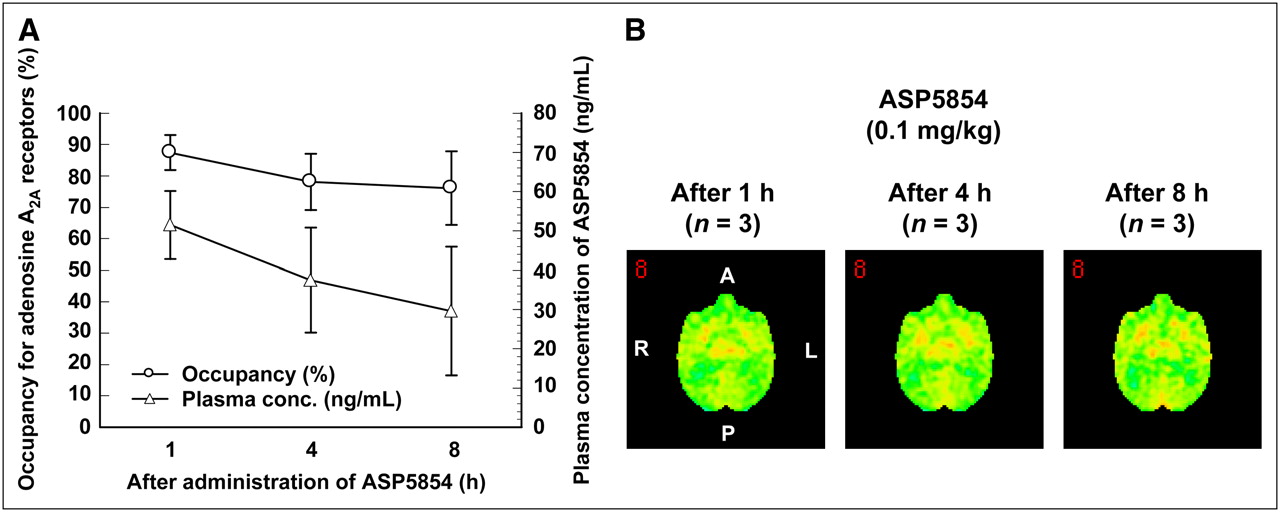
Correlation between occupancy for adenosine A2ARs and plasma concentration after administration of ASP5854 at dose of 0.1 mg/kg in rhesus monkeys. (A) Time-dependent changes of occupancy and plasma concentration of 0.1 mg of ASP5854 per kilogram. Values are mean ± SE of 3 animals. (B) Averaged PET images of 11C-SCH442416 from 3 animals, from 5 to 10 min after tracer administration. ASP5854 (0.1 mg/kg) was intravenously administered at 1, 4, or 8 h before 11C-SCH442416 administration. Images were anatomically standardized using NEUROSTAT and presented in cross-sectional images at striatum level.
Haloperidol-Induced Catalepsy
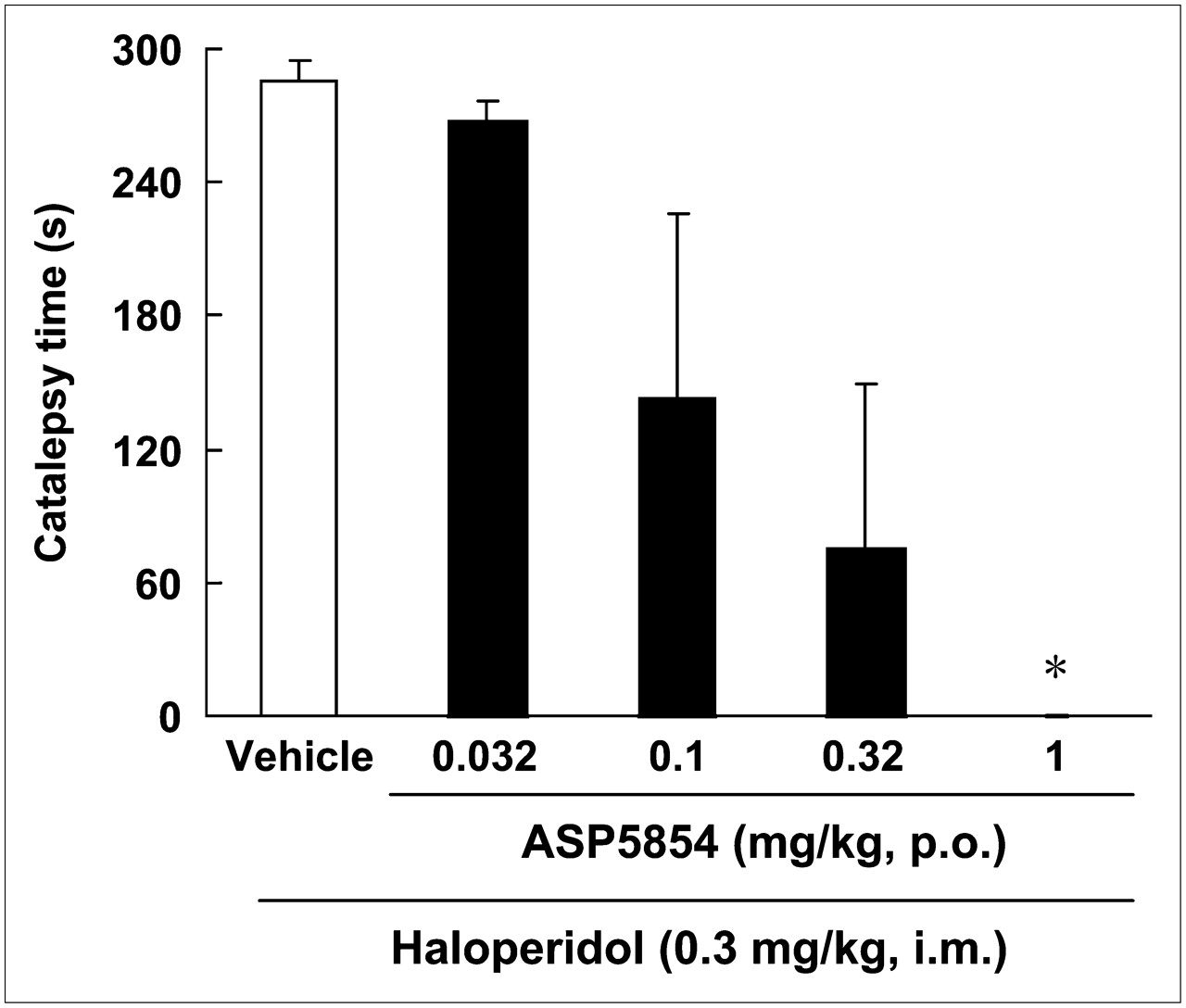
A 0.03 mg/kg dose of haloperidol induced cataleptic posture in all animals. One of the monkeys exhibited a strongly twisted torso. Oral administration of ASP5854 (0.032–1 mg/kg) reduced the incidence of onset-positive (incidence of onset-positive, 0.032 mg/kg, 4/4; 0.1 mg/kg, 2/4; 0.32 mg/kg, 1/4; and 1 mg/kg, 0/4) and caused dose-dependent reductions in the duration of catalepsy, with significant amelioration at 1 mg/kg (Fig. 4). The ED50 value was 0.1 mg/kg. The animals treated with 1 mg/kg of ASP5854 showed a freely moving without cataleptic or abnormal posture.
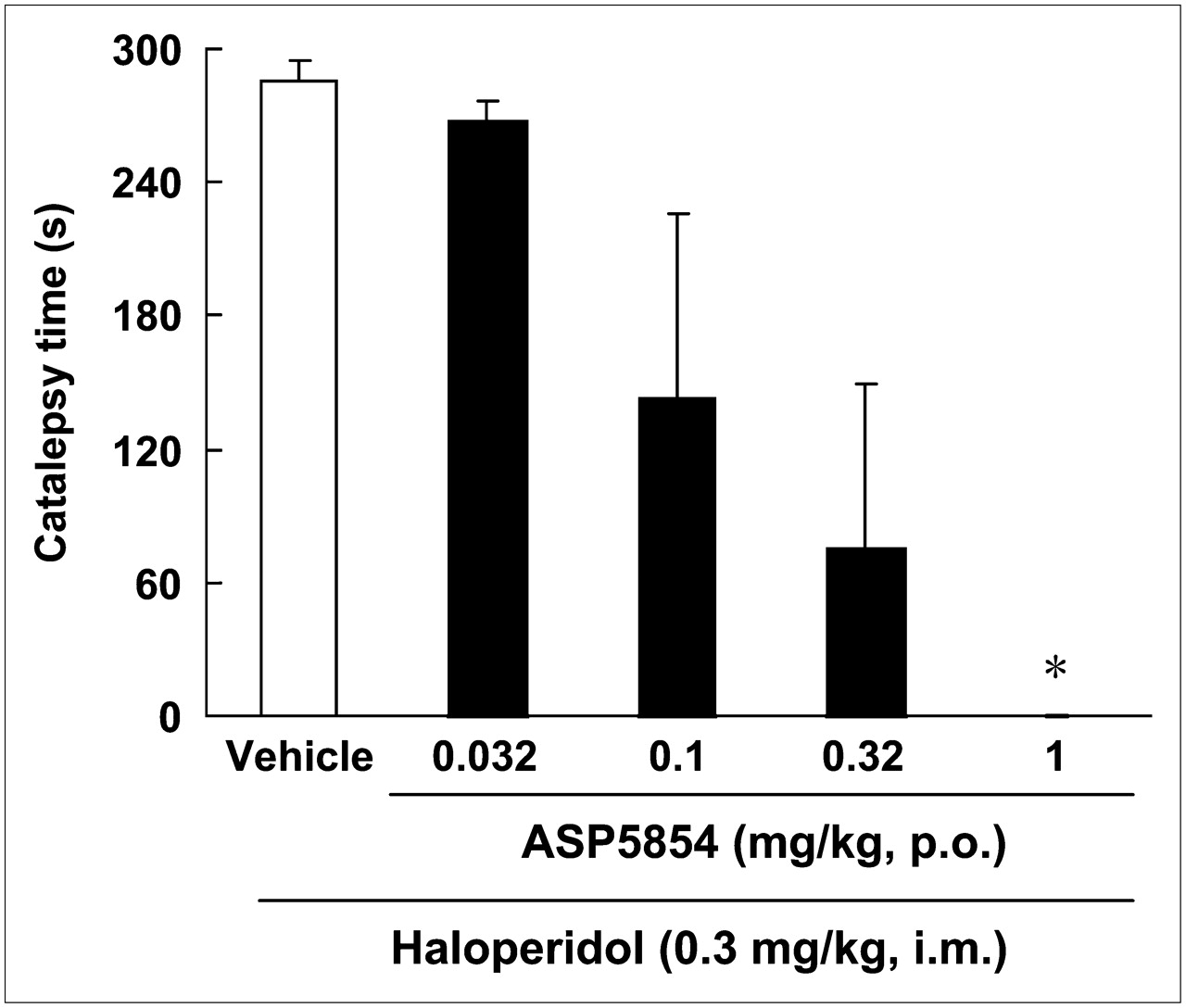
Effect of ASP5854 on haloperidol-induced catalepsy in rhesus monkeys. Catalepsy was measured 40 min after intramuscular injection of haloperidol. ASP5854 was orally administered 20 min before haloperidol injection. Each column represents duration of cataleptic posture and mean ± SE of 4 animals. ASP5854 suppressed haloperidol-induced catalepsy, and ED50 value was 0.1 mg/kg. *P < 0.05 (statistically significant compared with vehicle-treated group [by Kruskal–Wallis followed by Dunnett multiple comparison test]).
These 4 animals in the catalepsy test were given the anticataleptic ED50 value dose of 0.1 mg/kg (orally). The plasma concentration of ASP5854 was 97 ± 16.3 ng/mL at 1 h after the administration. This value corresponded to more than 85%–90% of occupancy for adenosine A2AR in the fitting curve of plasma concentration compared with receptor occupancy (Fig. 2).
DISCUSSION
The purpose of the present study was to identify the property of central adenosine A2AR occupancy by ASP5854 and the correlation between the receptor occupancy and antimotor impairment in nonhuman primates. ASP5854 has potent binding affinities for human adenosine A1 and A2A receptors without other molecular targets in the central nervous system (CNS) (13). This compound shows not only improvement of motor impairment in rodent models of PD but also neuroprotective effect on dopaminergic neuronal cell death by specific toxins and antidementia effects in animal models of cognition. It was also suggested that the antimotor-impairment effect of ASP5854 is primarily mediated by antagonism for central adenosine A2AR.
PET study is useful for the identification of the dynamic kinetics of drug to the target molecule in the organ without any surgical interventions. Adenosine A2ARs are abundant in the striatum, nucleus accumbens, and olfactory tubercle in the CNS area. High densities of adenosine A2AR in dopamine-rich regions have also been found in human, nonhuman primate, and rodent brains (27–29). In the present study, 11C-SCH442416 was used as a radiotracer to label brain adenosine A2ARs because ASP5854 has 2 molecular targets, adenosine A1 and A2A receptors, and the adenosine A1 receptors are known to distribute throughout the brain. 11C-SCH442416 showed a highly specific binding for A2ARs in the striatum with considerable signal-to-noise ratios consistent with the ratios found in earlier observations (20). ASP5854 dose-dependently displaced the binding of 11C-SCH442416 for adenosine A2ARs. This occupancy was long-lasting and not in accordance with reduction in the plasma concentration. This finding demonstrated that the dissociation of ASP5854 was relatively slow in the brain of primates and might be able to provide prolonged treatment by a single medication.
Haloperidol is one of the neuroleptic drugs and has high binding affinity for dopamine D2 receptors. This drug has been used for treatment of schizophrenia but often induces an extrapyramidal syndrome, including parkinsonism, as an adverse effect. In rats, haloperidol induces akinesia, muscular rigidity, and tremor of extremities, and these behaviors were simply assessed by catalepsy testing. Similar findings are also observed in nonhuman primates (30). Adenosine A2A antagonist and genetic disruption of adenosine A2ARs have anticataleptic activity. These effects were postulated to be caused by antagonistic interaction between adenosine A2A and dopamine D2 receptors at striatopallidal medium spiny neurons in the striatum (7). Thus, a catalepsy test is an adequate experiment to evaluate the adenosine A2A antagonism in vivo. Our rhesus monkeys showed cataleptic postures after haloperidol injection, a finding that is consistent with earlier observations (31). The incidence and duration of cataleptic posture produced by haloperidol injection was reduced by treatment of ASP5854 dose-dependently, a treatment result consistent with our previous observations in rodents. The antimotor-impairment effect of ASP5854 was induced by a dose of from 0.066 to 1 mg/kg, an ED50 value in various rodent motor-impairment models (13). In the present study with monkeys, the ED50 value was 0.1 mg/kg, suggesting that the effect of ASP5854 did not have significantly different results among species.
A pharmacokinetic study was conducted after behavioral assessment using the same animals to estimate a suitable occupancy ratio to induce the anticataleptic effect of ASP5854 in in vivo study. The dose of 0.1 mg/kg (anticataleptic ED50 value) of ASP5854 achieved a more than 85% occupancy ratio for striatal adenosine A2ARs. PET studies have recently been used to establish the correlation among dose level, receptor occupancy, and observed clinical effect in the discovery and development of CNS drugs (32). For instance, the central NK1 antagonist MK-0869 was shown to require more than 90% receptor occupancy to induce its antidepressant effect in nonhuman primates and humans (33). On the other hand, the antiemesis effect of MK-0869 was required to achieve 75% receptor occupancy for central NK1 receptors (33). Desirable receptor occupancy might be different depending on target symptoms, whereas it is the same molecular target in CNS. Therefore, whereas the correlation between effective plasma concentration and the receptor occupancy of ASP5854 might be reasonable, further studies using other animal models of motor impairment and also, ultimately, clinical studies would be required to estimate more accurately the antimotor-impairment effect of ASP5854.
CONCLUSION
The novel adenosine A1 and A2A antagonist ASP5854 was confirmed to be an orally active and brain-penetrable antagonist for adenosine A2ARs in nonhuman primates using the present PET study combined with a behavioral function study. It indicated that the anticataleptic effect of ASP5854 was achieved at more than 85% occupancy for adenosine A2A receptors. These findings suggest that the present methodology using PET might be useful for estimating the effective doses of adenosine A2A antagonists in humans, and ASP5854 might represent a novel treatment for PD.
Acknowledgments
We thank Dr. Raymond D. Price for providing helpful comments during the preparation of this manuscript and Takashi Sasagawa and Kaoru Sasaki for providing support of the behavioral experiment.
Footnotes
-
COPYRIGHT © 2008 by the Society of Nuclear Medicine, Inc.
References
- Received for publication February 7, 2008.
- Accepted for publication April 3, 2008.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.