


Abstract
Breast and endometrial cancers are the most common invasive malignancies in women, with more than 217,000 new diagnoses per year in the United States. These cancers are often classified into 2 subtypes based on the expression of the classical estrogen receptor. In this study, we describe a new structural class of neutral tridentate 99mTc(I)-estradiol-pyridin-2-yl hydrazine derivatives for potential use in breast and endometrial cancer imaging. Methods: The 99mTc(I)-estradiol-pyridin-2-yl hydrazine derivative was synthesized via the Sonogashira cross-coupling reaction and radiolabeled via the tricarbonyl approach. Radiochemical purity was assessed by high-performance liquid chromatography. Cell-binding studies were performed with human breast adenocarcinoma MCF-7 cells. The in vivo biodistribution of the 99mTc(I) derivative was evaluated in virgin female C57BL/6 mice in defined phases of the estrous cycle. Biodistribution and SPECT/CT studies were performed with mice bearing MCF-7 and primary human endometrial tumors. Results: Radiochemical analysis demonstrated that the postpurification purity of the 99mTc(I)-estradiol-pyridin-2-yl hydrazine derivative was ≥95%, with a specific activity of 99mTc of 47.5 TBq/mmol. Cell-binding studies yielded a dissociation constant (mean ± SEM) of 11 ± 1.5 nM. In vivo studies revealed that receptor-mediated uptake was present in all phases of the estrous cycle in reproductive organs and mammary glands but was highest during the diestrous phase of the estrous cycle. Despite high nonspecific uptake in the liver, significant receptor-mediated uptake was observed in target tissues and estrogen receptor–expressing tumors (0.67% for MCF-7 tumors and 0.77% for endometrial tumors). Tumor uptake was reduced by approximately 50% on coinjection with 17β-estradiol. Conclusion: We have characterized a novel neutral tridentate 99mTc(I)-estradiol-pyridin-2-yl hydrazine derivative for potential use in breast and endometrial cancer imaging. This study represents the first step on a path toward the design of estrogen-based Tc-labeled tracers with improved targeting and SPECT imaging characteristics.
- estrogen receptor
- small-animal SPECT imaging
- estrous cycle
- breast cancer
- endometrial cancer
- 99mTc-tricarbonyl
Over 250,000 new cases of breast, ovarian, and endometrial cancers were diagnosed in the United States in 2006 (1). Such cancers are often hormonally regulated and can be divided into 2 subtypes on the basis of whether or not tumor cells express the classical estrogen receptor (ER), ERα. Estrogen promotes cell proliferation and inhibits apoptosis through a complex signaling cascade resulting in transcriptional changes that may include the modulation of tumor suppressor function. The presence of ERα in approximately two thirds of breast cancers correlates with whether the tumors are estrogen dependent or independent (2) and represents one of the best prognostic factors in breast cancer because of the availability of antiestrogens such as tamoxifen and fulvestrant and, more recently, the aromatase inhibitors. With the recent characterization of a novel transmembrane ER, GPR30, in multiple cancer types, the exact causes and mechanisms underlying estrogen dependence and resistance in cancers may be more complex than previously appreciated (3–5). Nevertheless, it is clear that if a cancer can be detected at an early stage and characterized for ER status and metastatic state, then patients can display improved outcome with appropriate treatment (6,7). As a result, in the past decade, greater emphasis has been placed on the development of radioimaging approaches for breast and other cancers that are based on either SPECT or PET (8). Whole-body imaging is possible with PET and SPECT and therefore offers a noninvasive approach for assessing regional and disseminated cancers.
In 1986, a study involving 547 patients demonstrated the predictive value of determining estrogen-binding activity for the clinical behavior of breast cancer in women (9). Throughout the1980s, a variety of 18F-labeled estrogen derivatives were tested in animals and shown to demonstrate ER selectivity (10,11). The most successful 18F-labeled estrogen derivative, 16α-18F-17β-estradiol (18F-FES), has been evaluated clinically, with promising results for the imaging of estrogen-binding tumors and for predicting the responsiveness of breast tumors to antiestrogen drugs such as tamoxifen (12,13). The role of estrogen in endometrial carcinogenesis has been well documented (14,15), and successful applications of 18F-FES have been reported for endometrial and other gynecologic cancers (16). Subsequent efforts have been directed to developing methods for labeling estrogen imaging agents with the widely available and longer-lived radionuclide 99mTc for SPECT, through both pendant- and integrated-chelate approaches (17). Reports have described the generation and use of both steroidal and nonsteroidal estrogen derivatives labeled with 99mTc (SPECT) and 94mTc (PET) for ER imaging. However, the agents described to date have demonstrated suboptimal target tissue selectivity in vivo, possibly as a result of the high lipophilicity or rapid metabolism of these agents. The complex chemistry involved in the radiosynthesis of these compounds to obtain high yields and purities has further hampered their development for clinical use (18,19).
We recently described a new class of neutral tridentate rhenium-estradiol-pyridin-2-yl hydrazine derivatives (see Table 1 in the article by Ramesh et al. (20)). These compounds were biologically evaluated by competitive radiometric binding assays with 3H-estradiol to determine their relative binding affinities for ERα and ERβ. The relative binding affinities compared favorably with those of the best examples of previously reported estradiol tricarbonyl-Re(I) complexes (8,17,18). Binding to the alternate ER, GPR30, was also shown to be of high affinity. A functional assay based on the rapid receptor-mediated mobilization of intracellular calcium elicited by binding to ERα, ERβ, or GPR30 revealed that the alkyne-linked complex yielded the highest levels of receptor activation. It was also previously reported that the substitution of a 17α-ethynyl group reduced the affinity of estrogen derivatives for α-fetoprotein and sex steroid–binding protein, resulting in more favorable in vivo pharmacokinetics (21,22). In this report, we describe the detailed chemical and biologic evaluation of a 99mTc(I)-estradiol-pyridin-2-yl hydrazine derivative with an alkyne linkage for the diagnostic imaging of breast and endometrial cancers.
Stability of 99mTc(I)-Estradiol-Pyridin-2-yl Hydrazine Derivative
MATERIALS AND METHODS
Radiosynthesis of 99mTc(I)-Estradiol-Pyridin-2-yl Hydrazine Derivative
The 99mTc-tricarbonyl complex [99mTc(CO)3(H2O)3]+ was prepared by adding 3.7 GBq of freshly eluted Na-99mTcO4 to the Isolink kit reagent (Tyco Healthcare, Mallinckrodt) and heating the reaction mixture for 40 min at 100°C. The [99mTc(CO)3(H2O)3]+ complex was allowed to cool for 20 min on ice. Because of the acid sensitivity of the tertiary propargylic 17β-alcohol of the estradiol chelate, the alkaline mixture was neutralized to pH 7 with acetic acid. The estradiol-pyridin-2-yl hydrazine derivative was prepared as previously described (20). To generate the radiolabeled complex, the estradiol-pyridin-2-yl hydrazine derivative (10 μg, 21.7 nmol)) was added to the neutralized [99mTc(CO)3(H2O)3]+ complex, and the mixture was stirred for 2 h at room temperature. Heating was avoided to prevent β-elimination of the 17β-alcohol.
Inorganic impurities from the Isolink kit, aqua ions of 99mTc (if any) and excess ligand were separated by solid-phase extraction with C-18 SepPak Plus cartridges (Waters). Impurities and excess ligand were eluted with four 0.5-mL aliquots of 40% ethanol in water. Elution of the 99mTc-labeled estradiol-pyridin-2-yl hydrazine derivative was accomplished with 4 × 0.5 mL aliquots of 100% ethanol. Reverse-phase high-performance liquid chromatography (HPLC) was performed before and after purification to assess radiochemical purity with a reverse-phase C-18 column (JT Baker) with a γ-ram radiometric detector (INUS Systems Inc.). The injection volume was 100 μL, and the ultraviolet (UV) detector wavelength was 254 nm. The mobile phase (solvent A) consisted of 100% HPLC-grade ethanol whereas the stationary phase (solvent B) consisted of 100% HPLC-grade water. The elution gradient was 60%–70% solvent A over 10 min followed by 70%–100% over 15 min with a flow rate of 1 mL/min. Specific activity was determined from the integrated peak radioactivity (compensating for the elution profile of the 99mTc generator and the transient equilibrium between the parent and the daughter radionuclides) and the UV absorption with a calibration curve of known quantities of unlabeled compound.
Stability and Transchelation Studies
In separate tubes, the 99mTc derivative (final ligand concentration, 1 μM) was added to 900 μL of phosphate-buffered saline solution (PBS), mouse serum, 1 mM cysteine solution, 1 mM histidine solution, or 1 mM diethylenetriaminepentaacetic acid (DTPA) solution. The samples were incubated at 37°C and analyzed by HPLC and instant thin-layer chromatography after 1, 3, and 24 h.
Partition Coefficient Studies
Determination of log octanol/water partition coefficient (P(o/w)) values was performed by the shake flask method. To a solution containing 200 μL of octanol and 200 μL of water (obtained from a saturated octanol-water solution), 10 μL of 50 nM 99mTc(I)-estradiol-pyridin-2-yl hydrazine derivative was added. The resulting solution was vortexed at room temperature for 10 min and incubated at 37°C for 1 h. Aliquots (50 μL) were removed from the octanol and from the water phases, and the radioactivity measured in a Wallace Wizard 1480 automatic γ-counter.
Cell Culture
ERα/β- and GPR30-expressing human breast adenocarcinoma MCF-7 cells (HTB 22; American Type Culture Collection) were cultured in Dulbecco's minimal essential medium containing 10% fetal bovine serum, penicillin at 100 U/mL, and streptomycin at 100 μg/mL. Cells were grown as a monolayer at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
Receptor-Binding Studies
To evaluate ligand binding to the estrogen receptors expressed in MCF-7 cells, direct binding was performed with the 99mTc-labeled estradiol derivative. For saturation binding studies, 500,000 cells per tube in tissue culture medium were treated with increasing concentrations of the 99mTc-labeled estradiol derivative and incubated for 1 h at 37°C. The cells were pelleted and washed 3 times with 1 mL of PBS. The radioactivity associated with the final pellet was counted using a Wallace Wizard 1480 automatic γ-counter. To determine nonspecific binding, the cells were incubated with 10 μM 17β-estradiol. To establish the affinity of the corresponding Re-labeled derivative (20), which serves as an isosteric nonradioactive surrogate for the 99mTc-labeled derivative (19,23), cells were treated with a trace amount of the radioligand and competition was performed with increasing amounts of the Re-estradiol derivative. Binding data were analyzed with GraphPad Prism, Version 4 (GraphPad Software, Inc.).
Animal and Tumor Models
Animal use protocols were approved by the University of New Mexico Health Sciences Center Institutional Animal Care and Use Committee. To determine uptake during various stages of the estrous cycle, wild-type mature (8–10 wk old) female C57BL/6 mice (Harlan Inc.) were used. Estrous cycle was determined by cytologic examination of vaginal flushings (24). The mice used in this study were determined to be cycling by examination over 3–5 consecutive days before the day of the experiment.
Estrogen-dependent human breast adenocarcinoma MCF-7 tumors were generated by injecting 2–4 million MCF-7 cells subcutaneously in 8-wk-old female athymic NCr-nu/nu mice (NCI-Frederick). One day before injection of the cells, a 60-d release 17β-estradiol pellet (1.7 mg, Innovative Research of America) was implanted subcutaneously. After 6 wk, tumors ranging from 0.6 to 0.9 cm in diameter were observed; at this stage, ovariectomy was performed and the pellet was removed. After a 1-wk recovery period, biodistribution and imaging studies were performed.
The collection and use of human tumor specimens was approved by the Human Research Review Committees at the University of New Mexico Health Sciences Center. Fresh patient endometrial tumor specimens were rinsed with cold sterile PBS and grossly necrotic tissue was trimmed away. The tumor was minced and rinsed with PBS again. Approximately 100 mg of tumor suspended in 100 μL of Dulbecco's minimal essential medium was injected subcutaneously into 8-wk-old female athymic NCr-nu/nu mice (25). Xenograft tumors were harvested before any dimension reached 20 mm. The tumor used in this study was shown to be ER-positive by immunohistochemical staining (data not shown).
Biodistribution and SPECT/CT Studies
All mice were injected intravenously (tail vein) with the 99mTc-labeled estradiol derivative. To determine receptor specificity, 17β-estradiol (5 μg) was coinjected with the radiotracer. At the desired time point, the animals were sacrificed by CO2 euthanasia. After sacrificing the animals, organs were carefully removed and isolated to determine the biodistribution characteristics of the tracer. The organ samples were weighed and the corresponding localized radioactivity was measured using an automated γ-counter after verifying the counting efficiency with standards. The percentage injected dose per gram of tissue (%ID/g) was calculated by comparison with standards representing the injected dose per animal.
SPECT/CT studies were performed using a multi-pinhole SPECT/CT small-animal imager (Bioscan Inc.). Whole-body imaging studies were performed on anesthetized animals using 1.5%–2.0% isoflurane on a temperature-controlled bed. The images were reconstructed, fused and analyzed using the InVivoScope software program (Bioscan Inc.). All animal experiments were conducted in compliance with the guidelines and approved protocols established by the Institutional Animal Care and Use Committee.
Statistical Analysis
All numeric data were expressed as the mean of the values ± the SEM. Graphpad Prism, Version 4, was used for statistical analysis, and a P value of less that 0.05 was considered statistically significant.
RESULTS
Radiochemical Purity
Preparation of the [99mTc(CO)3(H2O)3]+ “semiaqua” intermediate was performed yielding a radiochemical purity of > 95% (n > 30). The starting aqua ion [99mTcO4]− had a retention time of 0.9 min and the intermediate semiaqua ion precursor had a retention time of 1.4 min. Over 85% of the [99mTc(CO)3(H2O)3]+ precursor was subsequently incorporated into the estradiol-pyridin-2-yl hydrazine derivative (Fig. 1). The prepurification radiochemical purity assessed by HPLC was greater than 85% (n > 25) (Supplemental Fig. 1A) (supplemental materials are available online only at http://jnm.snmjournals.org). The two resolved radiometric peaks (4.1 and 5.2 min) likely represent the two diastereoisomeric chelates of the final product based on similar chromatographic profiles of the corresponding Re-labeled derivatives (20). Based on the HPLC analyses (and taking into account the elution profile of the 99mTc generator and the transient equilibrium between the parent and the daughter radionuclides), the calculated specific activity of the final product was approximately 47.5 TBq of 99mTc/mmol. The radiochemical purity was further improved to ≥95% by solid-phase purification to remove excess ligand and unincorporated [99mTc(CO)3(H2O)3]+. Typical radiochemical yields after purification ranged from 60% to 80%.

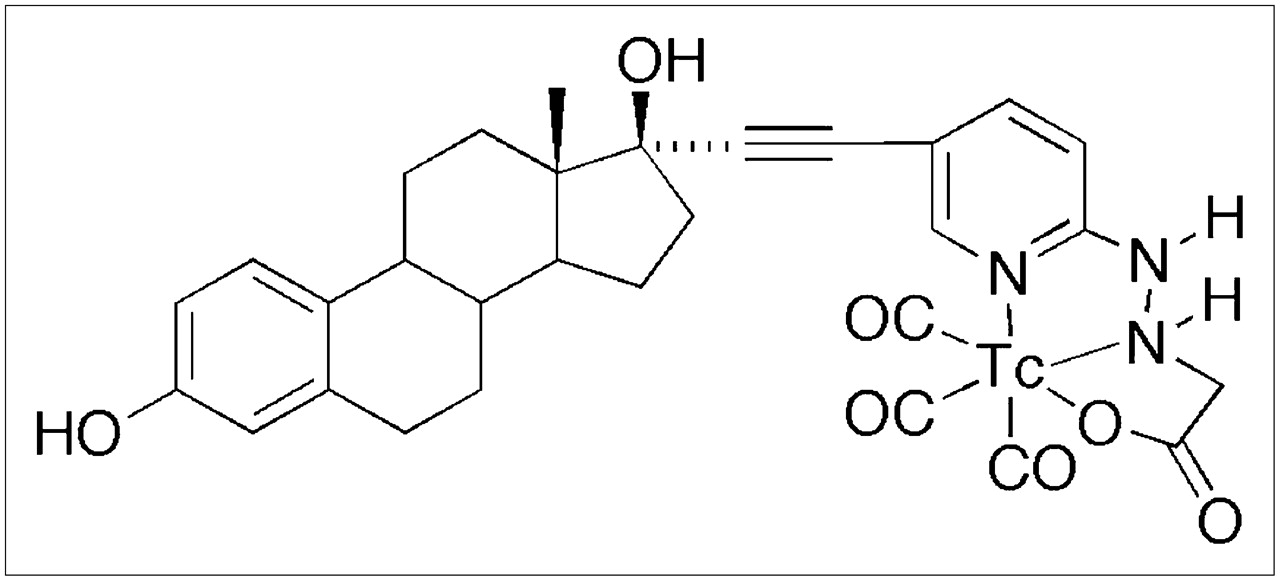
Chemical structure of the 99mTc(I)-estradiol-pyridin-2-yl hydrazine derivative possessing an alkyne linkage and prepared by the tricarbonyl approach.
Stability, Transchelation, and Partition Coefficient Studies
The final 99mTc(I)-estradiol-pyridin-2-yl hydrazine derivative demonstrated high stability in buffer and mouse serum after 24 h of incubation at 37°C (Table 1). Up to 30% transchelation was observed in the presence of a 1,000-fold molar excess of histidine or cysteine after 24 h of incubation at 37°C. The complex exhibited a log P(o/w) value of 3.9 ± 0.5 (n = 4) determined by shake flask method, compared with values of estradiol in the range of 3.3–5 (23).
Receptor-Binding Studies
Direct intact cell–binding studies were performed on ERα/β- and GPR30-expressing human breast adenocarcinoma MCF-7 cells. The 99mTc(I)-estradiol-pyridin-2-yl hydrazine derivative exhibited a dissociation constant (Kd) of 11 ± 1.5 nM with a calculated maximal number of binding sites (Bmax) of 1.3 × 104 ± 0.1 × 104 sites per cell (Fig. 2A), similar to previous estimates of the number of estrogen-binding sites in these cells (21). The inhibitory concentration of 50% (IC50) of the nonradioactive rhenium conjugate (15 ± 1.4 nM, dissociation constant of an inhibitor [Ki] = 10.3 nM) was similar to the Kd of the 99mTc-conjugate (Fig. 2B). These values also compare well with our previous determinations with the rhenium conjugate using competition of tritiated estrogen for purified ERα and a fluorescent estrogen for GPR30 in permeabilized cells where the relative binding affinities were 20% and 42%, respectively (20). The diastereoisomeric 99mTc(I)-estradiol-pyridin-2-yl hydrazine derivatives were separated by HPLC fractionation. Diastereoisomer A (retention time, 4.1 min) exhibited a Kd of 12 ± 2.9 nM, whereas diastereoisomer B (retention time, 5.2 min) exhibited a Kd of 7.7 ± 2.6 nM.

(A) Saturation binding curve with inset Scatchard plot of radioligand receptor-binding studies performed with 99mTc(I)-estradiol-pyridin-2-yl hydrazine derivative and MCF-7 human breast adenocarcinoma cells. 99mTc(I)-estradiol-pyridin-2-yl hydrazine derivative had Kd of 11.0 ± 1.5 nM and Bmax of 1.3 × 104 ± 0.1 × 104 sites per cell. (B) Competition binding curve from radioligand receptor-binding studies performed with 99mTc(I)-estradiol-pyridin-2-yl hydrazine derivative as radiotracer, corresponding nonradioactive Re-labeled derivative as competitor, and MCF-7 cells. Nonradioactive Re-labeled estradiol-pyridin-2-yl hydrazine derivative had IC50 of 15.0 ± 1.5 nM and calculated Ki of 10.3 nM. Values were determined from 3 independent experiments.
Role of Estrous Cycle
Receptor-mediated uptake was observed in reproductive organs and the mammary gland in all stages of the estrous cycle (Table 2). The highest uptake of the tracer in reproductive organs and the mammary gland was observed in diestrus and the lowest uptake during estrus (Table 2). At 3 h after injection, the uptake in the mammary glands was 0.22 ± 0.02 %ID/g in estrus and increased to 1.31 ± 0.13 %ID/g in metestrus and 3.11 ± 0.24 %ID/g in diestrus. A similar trend of increasing uptake levels from estrus to metestrus and diestrus was found in uterus and ovaries. However, the uptake of the radiotracer by the ovaries was not significantly different between estrus (0.60 ± 0.06 %ID/g) and metestrus (0.76 ± 0.04 %ID/g). As shown in Table 2, in bone, blood, and muscle, the uptake was not receptor mediated because there was no statistically significant difference in the values between unblocked and blocked treatments.
Uptake of 99mTc(I)-Estradiol-Pyridin-2-yl Hydrazine Derivative Throughout Estrous Cycle
Biodistribution in Non–Tumor-Bearing C57BL/6 Animals in Diestrus
The 99mTc-estradiol derivative demonstrated significant uptake in ER-expressing organs of interest with robust target tissue-to-muscle ratios in the range of 10:1 and moderate target tissue-to-blood ratios in the range of 1:1. In biodistribution studies, high liver uptake levels of 9.48, 8.34, and 7.84 %ID/g at 1, 3, and 24 h after injection, respectively, probably resulted from high compound hydrophobicity (Table 3). Low uptake in the kidney and bladder at 1 and 3 h after injection suggested excretion occurred slowly via urine. At 3 h after injection, approximately 5–10 %ID was excreted in the feces and less than 2 %ID was excreted in the urine. Urine and blood plasma was collected 3 h after injection and HPLC analysis were performed. No detectable radioactive metabolites were observed in the urine (Supplemental Fig. 1B) or plasma radiochromatograms (Supplemental Fig. 1C).
Time-Dependent Uptake of 99mTc(I)-Estradiol-Pyridin-2-yl Hydrazine Derivative
Biodistribution in Tumor-Bearing Animals
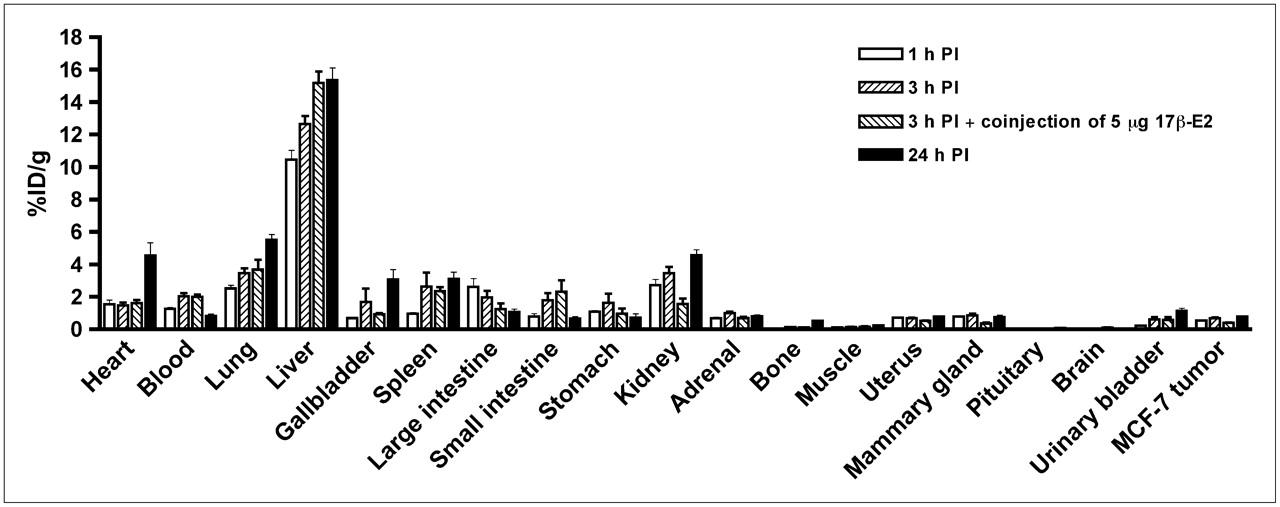
In tumor-bearing animals, in addition to receptor-mediated uptake in the estrogen-binding target organs, the 99mTc-estradiol derivative also displayed uptake in MCF-7 tumors and human primary endometrial tumors (Fig. 3). As in C57BL/6 mice, high liver uptake values were obtained in animals bearing MCF-7 tumors (Fig. 4) and human primary endometrial tumors. High levels of radioactivity were again found in the feces. The uptake in the blood for MCF-7 tumor bearing animals was 1.25, 2.03, and 0.81 %ID/g at 1, 3, and 24 h after injection, respectively. Similarly, uptake in the blood was 1.27, 1.59, and 1.00 %ID/g at 1, 3, and 24 h after injection, respectively. The target-to-muscle ratios were reasonably high to moderate, ranging from 3.5 to 7.05 over the period of time; however, the target-to-blood ratios were poor and ranged from 0.3 to 1.0 (Table 4). At 3 h after injection, the tumor-to-muscle ratio was 5.67 and the tumor-to-blood ratio was 0.35. The tumor-to-muscle and tumor-to-blood ratios were significantly reduced to 2.64 and 0.19, respectively, when blocking was performed with coinjection of 5 μg of 17β-estradiol.
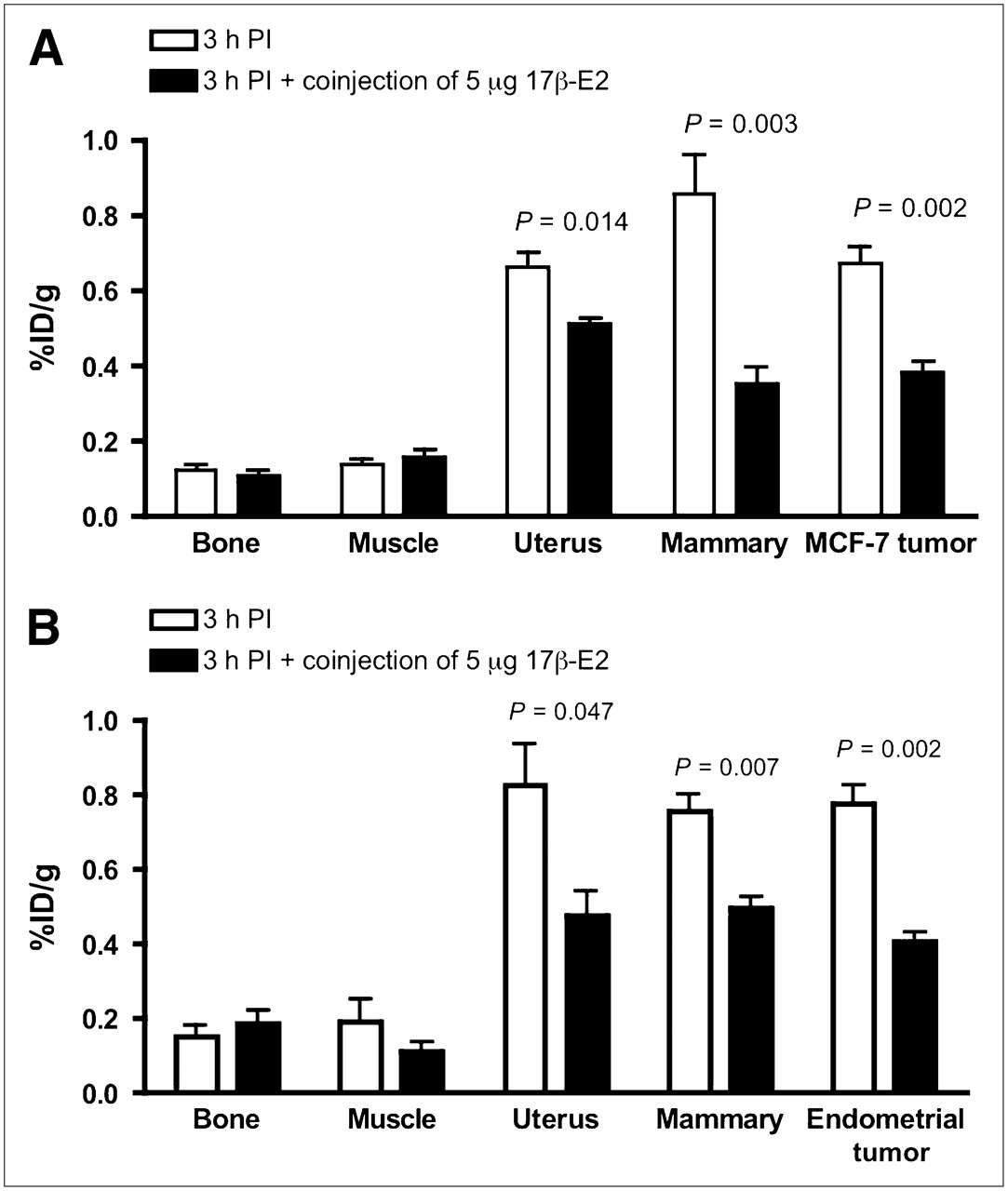
Receptor-mediated uptake of 99mTc(I)-estradiol-pyridin-2-yl hydrazine derivative in ovariectomized female athymic NCr-nu/nu mice bearing MCF-7 human breast adenocarcinoma tumors (A) or human primary endometrial tumors (B). Uptake values are expressed as %ID/g and were determined from biodistribution studies. Data represent mean ± SEM from at least 3 determinations. PI = postinjection.
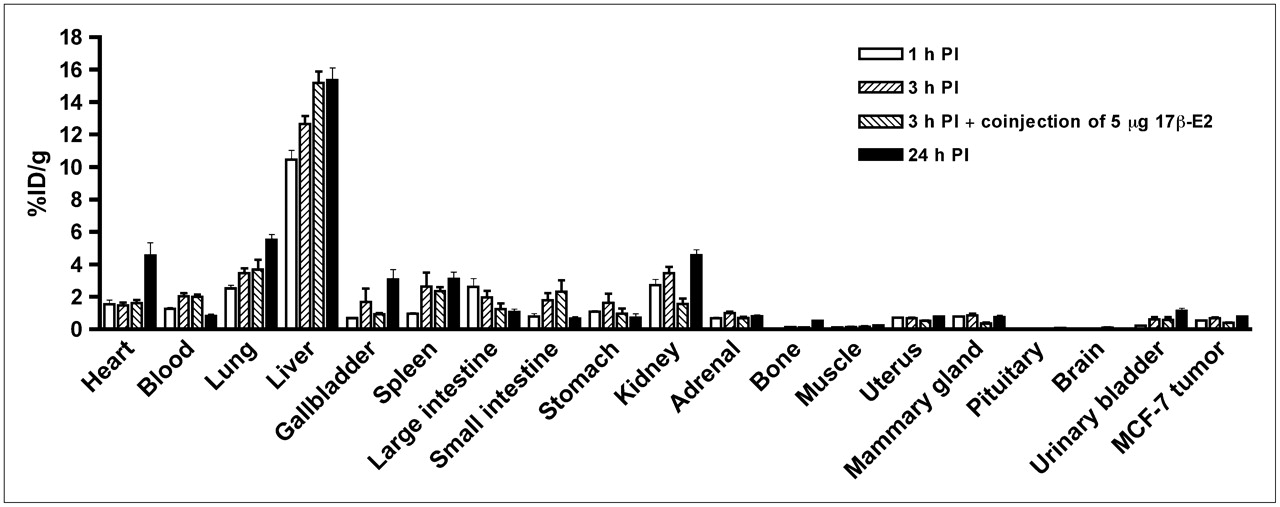
Uptake of 99mTc(I)-estradiol-pyridin-2-yl hydrazine derivative in selected organs of ovariectomized female athymic NCr-nu/nu mice bearing MCF-7 human breast adenocarcinoma tumors. Biodistribution data were obtained at 1, 3, and 24 h after injection. All uptake values are expressed as %ID/g. Data represent mean ± SEM from at least 4 determinations. PI = postinjection.
Target-to-Muscle and Target-to-Blood Ratios of 99mTc(I)-Estradiol-Pyridin-2-yl Hydrazine Derivative
Imaging Studies
Imaging studies were performed after injecting 18.5 MBq of the 99mTc(I)-estradiol-pyridin-2-yl hydrazine derivative via the tail vein. Whole-body imaging studies at 60 s per projection were performed under anesthesia with a temperature-controlled bed (36°C–38°C). In this study, very low counts were obtained in tumors and they were only visualized once the image threshold was reduced to 2% maximum intensity. Whole-body imaging studies also revealed high uptake in the liver and intestines (i.e., feces) (82.8% ± 1.9 %ID combined). Uptake in the MCF-7 tumor (3 h after injection) was relatively low (0.078% ± 0.010 %ID) when compared with the background. A focused study at 600 s per projection was performed postmortem for better visualization of the tumor to obtain higher counts. In this study, tumor visualization was better without significantly reducing the %ID scale (Fig. 5). In the reconstructed coregistered SPECT/CT maximum-intensity-projection image (Fig. 5A) and the reconstructed coregistered SPECT/CT sagittal slice (Fig. 5B), the radioactivity in the tumor and the liver can be visualized, whereas in the reconstructed coregistered SPECT/CT transverse slice (Fig. 5C) the localization of radioactivity can be visualized without any significant background.
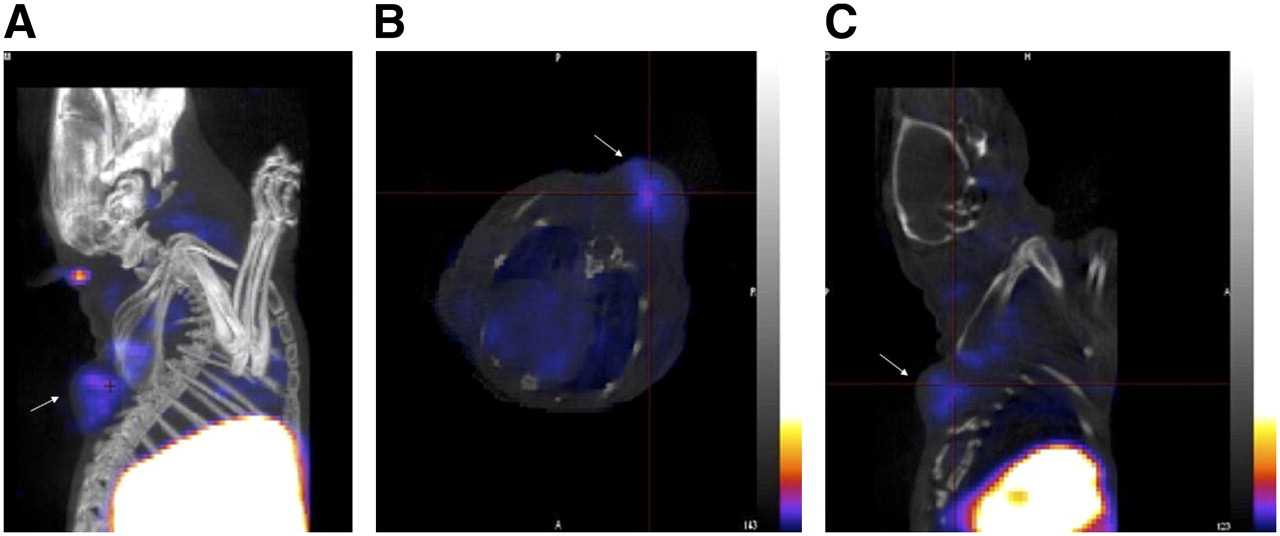
(A) Reconstructed coregistered maximum-intensity-projection SPECT/CT image. (B) Reconstructed coregistered transverse SPECT/CT slice image. (C) Reconstructed coregistered sagittal SPECT/CT slice image. In all images, the tumor is indicated by an arrow. Images were acquired from ovariectomized female athymic NCr-nu/nu mice bearing MCF-7 human breast adenocarcinoma tumors and injected with approximately 18.5 MBq of 99mTc(I)-estradiol-pyridin-2-yl hydrazine derivative via tail vein. Images were acquired postmortem at 3 h after injection. For SPECT image, focused scan at 600 s per projection was used; CT image was acquired with 180 projections (pitch of 1.5 and energy of 45 kVp). Energy window for SPECT acquisition of 99mTc was set at 140 ± 14 keV. Images were reconstructed, fused, and analyzed with InVivoScope software program. In these representative SPECT images, maximum intensity of color scale was set to 25%.
DISCUSSION
Advances in molecular and cellular biology are transforming our understanding of basic physiology and pathology; similar advances in molecular imaging technologies now permit dynamic and quantitative studies in vivo with minimal invasiveness. In this study, we have developed a novel γ-emitting estradiol derivative and used it to investigate the interaction with estrogen receptors in defined stages of the estrous cycle in normal animals and also to evaluate its effectiveness in imaging mouse xenograft breast and endometrial tumors with SPECT.
Because of the desirable imaging characteristics of 99mTc, several groups have prepared 99mTc-labeled estradiol derivatives as breast cancer imaging agents. Most of the previously described 99mTc-labeled estradiol derivatives were modified at the 7α- and 17α-positions for incorporation of 99mTc (18,19). None of these reported steroidal analogs was successful as ideal imaging agents, because of either low binding affinity or the difficulty in preparing 99mTc-labeled derivatives with high specific activity and yields. However, it was observed that the analogs with the best characteristics were those substituted at the 17α-position. Based on our previous characterization of 17α-substituted estradiol derivatives with respect to receptor binding and cell activation (20), we surmised that the use of an ethynyl group for linkage at the 17α-position would enhance receptor binding as well as cell permeability. Incorporation of a pyridyl moiety would allow for the incorporation of a [99mTc(CO)3(H2O)3]+ group for production of an imaging agent. As a result, we prepared a neutral tridentate 99mTc(I) estradiol pyridin-2-yl hydrazine derivative using the tricarbonyl approach.
Because of the acid sensitivity of the tertiary propargylic 17β-alcohol of estradiol, the alkaline mixture was neutralized with acetic acid rather than HCl and heating was avoided to prevent elimination. Direct saturation binding studies were performed with the radioactive 99mTc-labeled derivative, and competition binding studies were performed with the nonradioactive Re-labeled derivative on human breast adenocarcinoma MCF-7 cells. Performing receptor-binding studies on whole cells with steroidal analogs poses challenges related to ligand equilibrium, the lipophilic nature of the ligand, estimation of free ligand, the processes of receptor activation and degradation, and the presence of multiple estrogen receptor types, including GPR30 (26). As a result, careful consideration is required for data interpretation as one can easily over- or underestimate the Kd. The 99mTc(I) estradiol pyridin-2-yl hydrazine derivative demonstrated a Kd of 11 ± 1.5 nM on MCF-7 cells, much higher than typically reported values of estradiol itself (∼0.1–1 nM (27)) but comparable to the affinity of estrogen for GPR30 (∼6 nM (26,28)). Furthermore, this Kd value is consistent with the affinities previously determined for the Re-labeled derivative (20). Because chelate diastereoisomers demonstrated similar binding affinities in vitro, in vivo studies with separated diastereoisomers were not performed. For in vivo evaluation, a mixture of diastereoisomers was injected in mature (8–10 wk old) female C57BL/6 mice in defined stages of estrous cycle.
In studies with radiolabeled estrogen derivatives, it has been observed that the maximum uptake in the uterus is achieved during the diestrus phase (29,30); our finding that the highest uptake of 99mTc-labeled estrogen derivative in uterus, ovaries, and mammary gland occurs during diestrus is consistent with these observations. Circulating estrogens are low during estrus, metestrus, and early diestrus. Then, toward late diestrus and into proestrus, just before ovulation, levels rise significantly. Available estrogen receptor concentration is determined by circulating estrogens, being reduced by ligand binding, but also being upregulated by estrogen-mediated receptor synthesis (31). Thus, at diestrus the combined conditions of minimal estrogen levels and maximum estrogen receptor levels favor radiotracer uptake.
Another factor that may influence radiotracer uptake is hormonal regulation of the vasculature during the reproductive cycle. Blood flow to the ovaries but not to the uterus is increased during estrus (32). However, uptake in the ovaries during estrus and metestrus was not significantly different (33). In addition, blood flow is not uniform in the left and right ovaries and therefore may affect experimental outcomes. In a recently published clinical study, endometrial standardized uptake values determined by 18F-FES PET were significantly higher in the proliferative phase than in the secretory phase (34), highlighting the influence of menstrual cycle and endogenous estrogen levels in radiotracer uptake in humans.
Less is known about changes in the normal vasculature of the mammary gland; however, it has been observed that vascular permeability (but not blood flow or vascular density) is highest at diestrus (35). Importantly, the increase in vascular permeability at diestrus correlates with increased breast cancer growth rate and postresection metastasis (35). Thus it is possible that the 14-fold increase in radiotracer uptake at diestrus is related to changes in vascular permeability. It would be worth investigating the possibility that elevated ligand availability during the luteal phase in women is linked to reduced 5-y disease-free survival in women with breast cancer who undergo mastectomy (along with tamoxifen and oophorectomy) in the follicular phase as compared with the luteal phase (36). It should also be noted that not all human studies have found a difference in disease-free survival linked to the menstrual cycle (12).
The major goal for cancer imaging is accurate disease characterization through the application of functional and molecular imaging studies. 18F-FES PET cancer imaging has been immensely valuable to clinical oncologists for staging and visualizing primary and metastatic carcinomas (16,37). The supplemental molecular characterization and receptor-expression assessment of the tumor has often assisted in determining endocrine therapy options (12,13,38). As an alternative to PET, we wanted to evaluate the utility of a 99mTc(I)-estradiol-pyridin-2-yl hydrazine derivative in SPECT of breast and endometrial cancers. In previously published studies, problems encountered in developing a neutral Tc-estradiol derivative suitable for imaging of tumors were associated with excessive lipophilicity (i.e., logP values of >5 (23)) resulting in high liver and intestine uptake, low tumor-to-blood ratios, and relatively low uptake in the tumor compared with that in the background (17–19). In the present study, we encountered similar problems despite the fact that the logP value of our tracer was not significantly different from that previously reported for estradiol itself (3.9 vs. approximately 3.7 (23), respectively). From biodistribution studies, uptake of the radiotracer in the tumor was receptor-mediated based on the significantly lower uptake on coinjection with excess17β-estradiol. Care must be taken while performing receptor blocking experiments with high doses of 17β-estradiol as estrogen alters vascular physiology, blood flow and receptor expression levels (39). Consequently, when the blocking dose was injected 1 h before the injection of the radiotracer, we observed higher uptake in organs such as liver and lungs.
In this study, we chose the MCF-7 tumor model as representative of estrogen-dependent carcinomas; however, this model requires estrogen supplementation, which creates additional technical challenges involving ovariectomy and estrogen supplementation with pellets. For example, when we performed radiotracer studies 3 d after removing the estrogen pellet and the ovaries, the uptake in target organs was not blocked by excess ligand, likely attributable to high endogenous levels of estrogen. It is also known that estrogen supplementation and pregnancy can elevate the levels of sex steroid–binding protein and α-fetoprotein, which result in lower target-to-blood ratios (30). Our studies revealed that a period of at least 8 d after pellet and ovary removal was required to obtain significant results, although oophorectomy is not typically an option in the clinical setting. The low level of tracer uptake in tumor compounded with a high background resulted in poor whole-body SPECT/CT images at 60 s per projection. The image quality was greatly improved when a focused study was performed at 600 s per projection (Fig. 5). As a proof of principle, we have demonstrated that the tracer localizes in the tumor; however, such long acquisition time studies may not be feasible in clinical settings.
Currently, there are no readily available and easily synthesized SPECT agents for assessing reproductive cancers expressing estrogen-binding activity. Despite the challenges and drawbacks, newer 99mTc-based estradiol derivatives have been reported, including the one in this study. However, to date, all of them have failed to provide an alternative to 18F-FES PET, mainly attributable to low tumor uptake compounded with high background levels. To overcome the problems posed by such steroidal analogs, we propose the development of neutral nonsteroidal analogs that would specifically bind to each estrogen receptor subtype (ERα, ERβ, and GPR30) with high affinity (28,40).
CONCLUSION
In conclusion, in the present study, the highest levels of target uptake were observed during diestrus consistent with differential expression of ER and estrogen levels during the estrous cycle. Although the 99mTc(I)-estradiol-pyridin-2-yl hydrazine derivative showed encouraging results in standard biodistribution studies, as it demonstrated receptor-mediated uptake in normal target organs as well as human breast adenocarcinoma MCF-7 tumors and primary human endometrial tumors, further structural modifications are needed to optimize these compounds for improved imaging characteristics.
Acknowledgments
This study was supported by NIH grants CA116662 and CA127731, NIH grant SCORE GM08136, NIH grant MH074425, the University of New Mexico Cancer Research and Treatment Center (NIH P30 CA118100), the New Mexico Cowboys for Cancer Research Foundation, the Oxnard Foundation, and the Stranahan Foundation. Images in this article were generated at the Keck-UNM Small Animal Imaging Resource established with funding from the W.M. Keck Foundation. The technical assistance of Tamara Anderson and Kamalika Nag is greatly appreciated. We thank Dr. Robert Atcher (Biosciences Division, Los Alamos National Laboratory) for his valuable insights and Dr. Mary Dyszlewski (Tyco Healthcare, Mallinckrodt) for providing the Isolink kit.
Footnotes
-
COPYRIGHT © 2008 by the Society of Nuclear Medicine, Inc.
References
- Received for publication October 23, 2007.
- Accepted for publication February 18, 2008.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}