


Abstract
The transfer of peptide sequences identified by screening of phage-displayed libraries to clinical application is often difficult. This study investigated whether coupling of a new peptide, FROP-1, to the chelator 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) resulted in structural restriction and, consequently, improved binding and stability. Methods: The peptide FROP-1 was coupled to the chelator DOTA and labeled with 111In. The structural changes caused by the addition of the chelator were determined by circular dichroism. The properties of this modified peptide were investigated in in vitro binding assays and monitored for kinetics, competition, and internalization as well as serum stability. A cell-type binding profile was established and the in vivo biodistribution was evaluated in a nude mouse model. Results: When compared with the free peptide without chelator, FROPDOTA revealed different cellular uptake kinetics, reaching a maximum at 2 h in vitro. The cells completely accumulated the tracer, and competition experiments revealed that 99.4% (FRO82-2 cells), 98.6% (MCF-7 cells), or 99.3% (average for 3 primary head and neck tumor cell lines) of tracer accumulation could be suppressed, revealing the specificity of this process. The internalization kinetics determined in MCF-7 cells supported this finding: After an incubation time of 180 min, the major fraction of FROPDOTA was trapped intracellularly. Serum stability experiments revealed an increase in stability due to the chelator, with a half-life of 71 min. Circular dichroism measurements indicated a fixed α-helix structure of FROPDOTA representing a strong change in secondary structure. In competition binding experiments, the binding constant (KD) to FRO82-2 cells was determined to be 494 nM. Despite this avid binding affinity, the binding kinetics were found to be too slow to induce an uptake in vivo before clearance. Consequently, the biodistribution revealed a rapid renal and hepatobiliary clearance, with blood levels dropping from 5.48 ± 0.26 %ID/g (percentage injected dose per gram) 5 min after injection to 0.77 ± 0.15 %ID/g at 135 min after injection. Conclusion: This study revealed that peptides that are identified by display techniques may be underrated. Careful alteration of their structure will permit going beyond the possibilities that the limited pool of naturally occurring peptides provide for tumor targeting.
Naturally occurring peptides—in particular, small regulatory peptides—have been successfully used for the development of tracers for targeting purposes (1). The most prominent examples of peptide tracers are diethylenetriaminepentaacetic acid (DTPA)-octreotide and [DOTA0,Tyr3]-octreotide (DOTATOC) (DOTA is 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) that were developed on the basis of the somatostatin receptor-binding octreotide. 111In-DTPA-octreotide, known as OctreoScan (Mallinckrodt, Inc.), was the first commercially available radiopeptide and is still intensively used. DOTATOC allows imaging and therapy of somatostatin-expressing tumors (2,3). However, it would be advantageous to expand the options that natural peptides offer for the tumor targeting (4) by the identification of novel, artificial peptides. The identification and characterization of novel peptides that bind to cell-surface receptors overexpressed on tumors can be achieved by the screening of phage-display libraries (5). However, the expectations for this technology have not been met for in vivo applications.
The affinity of peptides identified by phage display is in general high enough and, therefore, 2 major reasons may cause this failure: (a) the rapid degradation of free peptides by peptidases and (b) a secondary structure that differs between the free peptide and the phage-bound peptide. The generation of structural constraints has to be considered for peptides that are identified by phage-display technologies because within the screening process the peptide is covalently attached to the large phage scaffold (6). There is no information about the structural interactions of phage-bound peptides with their scaffold available. This lack of a basis for a “rational design” prompted us to follow indications of an extensive influence of the secondary structure. Measurements of the circular dichroism (CD) provide specific structural information of peptides in solution. The DOTA conjugation (a) induces a strong change of the secondary structure and (b) results in a conjugate with improved characteristics such that we assume the interaction with the chelator resembles the interaction with the phage. However, this is only an assumption.
One major advantage of peptide tracers is their small size, allowing rapid distribution and clearance. Consequently, a peptide that is intended for use as a diagnostic or therapeutic tracer has to be used in the free form without the phage carrier protein. In this form the interaction with the large scaffold that influences both the stability and the conformation of the peptide is absent. Despite the fact that a large number of peptides with high selectivity in vitro have been determined, none of the phage-derived peptides has yet entered the stage of clinical application. Therefore, the translation of the enormous targeting potential to pharmacologically valuable structures is a major task after identification of the novel tumor-binding structures (7). For nuclear medicine applications, the straightforward solution for this is the attachment of the chelator to the C-terminal end, to stabilize the peptide against enzymatic degradation and to induce structural constraints that resemble those in the phage-bound structure.
In the preceding article, the isolation and characterization of FROP-1, a novel peptide that enables the selective targeting of thyroid carcinoma and a series of other tumors, was described (8). The beneficial properties of FROP-1—in particular, its highly selective binding, suggesting a unique tumor specificity—are hampered by a relatively rapid degradation that results in a relatively quick release from the cells. In this article, we describe the synthesis and preclinical study of FROPDOTA (H-EDYELMDLLAYLK(DOTA)-NH2), a chelate-modified derivative of FROP-1, and the functional effects of the chelate on the peptide properties caused by structural restrictions.
MATERIALS AND METHODS
Cell Lines
The human thyroid follicular carcinoma cell line FRO82-2 (University of California, Los Angeles, CA) was cultivated in RPMI 1640 medium containing Glutamax, plus 10% fetal calf serum (FCS) (Invitrogen Karlsruhe), 0.15% sodium bicarbonate, 1.5 mM sodium pyruvate, 1.5× nonessential amino acids (all Invitrogen), and 25 mM N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (HEPES). The human anaplastic thyroid carcinoma cell line SW1736 (obtained from Nils-Erik Heldin, Uppsala, Sweden) and the human colorectal carcinoma cell line HCT-116 (European Collection of Cell Cultures) were cultured in RPMI 1640 medium with Glutamax, containing 10% FCS and 25 mM HEPES. The human breast cancer cell line MCF-7 (Tumorbank; German Cancer Research Center) and the human squamous cell carcinoma cell lines HNO 97, HNO 210, and HNO 223 (obtained from Dr. Christel Herold-Mende, University Hospital Heidelberg, Germany (9)) were cultured in Dulbecco's modified Eagle medium with Glutamax, containing 10% FCS and 25 mM HEPES. The human HPV-16 immortalized keratinocyte cell line HPV-16-GM (obtained from Dr. Pascal Tomakidi, University Hospital Heidelberg, Germany) was cultured in KGM-2 medium with supplements (Promocell). Human umbilical vein endothelial cells (HUVECs) were isolated as described in the literature (10). HUVEC cultivation was performed on 1% gelatin-coated cell culture flasks using medium 199 (Invitrogen) containing 20% FCS, 2 mM glutamine, 100 IU/mL penicillin, 100 IU/mL streptomycin, and 2 ng/mL basic fibroblast growth factor (Roche Diagnostics). All cell lines were cultivated at 37°C in a 5% CO2 incubator.
Peptide Synthesis
FROPDOTA (H-EDYELMDLLAYLK(DOTA)-NH2) was obtained by solid-phase peptide synthesis using Fmoc-chemistry on a Rink amide resin (Rapp Polymere). Nα-Fmoc amino acids with the following side-chain protecting groups were used: Asp(t-Bu), Glu(t-Bu), and Tyr(t-Bu). DOTA was inserted using the preformed lysine conjugate Fmoc-Lys(tris-t-BuDOTA)-OH (11). The peptide chain was constructed in an automated Ecosyn P peptide synthesizer (Eppendorf). The reaction cycle consisted of a 2-fold decoupling (5 and 20 min) with 33% piperidine in N,N,-dimethylformamide and 30-min coupling with 4 equivalents of the 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate–activated Fmoc-protected amino acid. Cleavage from the resin was performed with 95:2.5:2.5 trifluoroacetic acid (TFA)/H2O/triisopropylsilane for 1 h at room temperature and subsequent precipitation with cold ether. The precipitate was dried and left in neat TFA for 12 h. Purification was accomplished by reversed-phase high-performance liquid chromatography (RP-HPLC) on a Chromolith SemiPrep 100 × 10 mm column (Merck). The purified peptide was characterized by analytic HPLC and mass spectrometry.
Peptide Labeling
The radiolabeling was performed by complexation with 111In-chloride. One microliter of a 10−3 M solution of FROPDOTA in 50% dimethyl sulfoxide in water was mixed with 50 μL of sodium acetate buffer (pH 5), and 10 MBq 111InCl3 (Tyco Healthcare Neustadt) were added. The solution was heated at 95°C for 25 min. A quality control of the radiolabeled peptide performed by HPLC on a monolithic RP-HPLC 100 × 4.6 mm column (Merck) using 0.1% TFA in water and 0.1% TFA in acetonitrile as eluents showed yields of >95%.
CD
Samples at a concentration of approximately 100 μg/mL were measured in a solution of distilled water containing 10% trifluoroethanol (TFE) to improve solubility. (At this low level, the effect of TFE on the secondary structure of most peptides tends to be negligible.)
CD spectra were measured on a Jasco J-710 automatic recording spectral polarimeter coupled with a Jasco PFD-35OS Peltier temperature control device. The instrument is routinely calibrated with 0.05% β-androsterone in dioxane. CD spectra were taken over the range 190−240 nm at a scanning speed of 5 nm/min and a 4.0-s time constant. The spectra presented are the result of 4-fold signal averaging with a similarly signal-averaged baseline subtracted. Spectra measured in millidegrees were converted to θMR for curve fitting and secondary structure analysis. Residual noise was removed using a Fast Fourier Transform program. For the analysis of the secondary structure, the processed spectra were fitted using the computer program PEPFIT (12). This program is specifically designed for the analysis of the secondary structure of peptides, as opposed to globular proteins, and is based on the known CD spectra of model peptides.
In Vitro Binding Experiments
FRO82-2 cells (400,000 cells) or MCF-7 cells (200,000 cells) were seeded into 6-well plates and cultivated for 24 h. The medium was replaced by 1 mL fresh medium (without FCS). 111In-FROPDOTA was added and incubated for 5, 10, 15, 30, and 60 min and 2, 4, 6, 24, 30, 48, 54, and 72 h. A control experiment in which no cells were seeded into the wells was conducted as described.
When using the competitor, unlabeled FROP-1 (without chelator) (10−4 M) was added to the incubation media immediately before the 111In-labeled peptide was added to the cell culture (1–2 × 106 cpm/well), and incubation was performed for the appropriate incubation times varying from 1 min to 4 h. The cells were washed 3 times with 1 mL phosphate-buffered saline (PBS) and subsequently lysed with 0.5 mL of 0.3 M NaOH. Radioactivity was determined with a γ-counter and calculated as the percentage applied dose per 106 cells. If bovine serum albumin (BSA) was used as the blocking agent, it was added to a final concentration of 1% in the medium without FCS.
To determine the concentration-dependent ability of FROP-1 to inhibit the binding of FROPDOTA, 400,000 subconfluent FRO82-2 cells were coincubated with 111In-FROPDOTA and FROP-1 at concentrations of 10−4 to 10−8 M. After an incubation time of 180 min at room temperature, the incubation medium was removed, and the cells were washed 3 times with 1 mL PBS and subsequently lysed with 0.5 mL of 0.3 M NaOH. Radioactivity was determined with a γ-counter and calculated as the percentage applied dose per 106 cells. Specific binding was defined as total binding minus nonspecific binding.
Cell Counting
Immediately after the completion of an in vitro binding experiment, the number of cells in 3 reference wells was determined. The wells were washed with PBS before 1 mL ethylenediaminetetraacetic acid (EDTA)-PBS solution (1 mM) was added to detach the cells. Cell aggregations were disrupted by pipetting, and the 1-mL cell suspension was added to 20 mL of cell counting liquid (Coulter Isoton II Diluent; Beckman-Coulter). Five hundred microliters of the suspension were counted 3 times in a cell counter (Coulter-Counter; Beckman-Coulter) and multiplied by 42 to obtain the cell number per well, and the mean value of the 3 reference wells was calculated.
Internalization Experiments
Internalization experiments were performed as described in the literature (7,13). Subconfluent cell cultures of the target cell lines were incubated with 111In-labeled peptide for 10, 60, and 180 min at 37°C and at 4°C. Cellular uptake was stopped by removing the incubation medium from the cells and washing 3 times with 3 mL of ice-cold PBS. To remove the radioactivity bound on the surface of the cells, the cells were incubated twice with 1 mL glycine-HCl in PBS (50 mM, pH 2.8) for 5 min at room temperature. The cells were subsequently washed with 3 mL of ice-cold PBS and lysed with 0.5 mL of 0.3 M NaOH. The surface-bound and the internalized radioactivities were measured in a γ-counter and calculated as the percentage uptake per 106 cells. To determine the specificity of the radioligand internalization, cells were also incubated in the presence of unlabeled FROP-1 at a concentration of 10−4 M.
Determination of Metabolic Stability
111In-FROPDOTA (4 MBq) dissolved in 200 μL was mixed with 500 μL human serum and incubated at 37°C. Samples were taken after 1, 2, 5, 15, 30, 60, and 120 min of incubation and mixed with acetonitrile to precipitate the serum proteins on ice. The suspension was centrifuged for 2 min at 13,000 rpm, and the supernatant containing the peptide was transferred into a second vial. This supernatant sample was recentrifuged for 10 min at 13,000 rpm, and the degree of degradation was determined by RP-HPLC on an Agilent 1100 series liquid chromatograph. RP-HPLC runs were performed using a linear A–B gradient (0% B to 100% B in 5 min) at a flow rate of 4 mL/min, where solvent A is 0.1% aqueous TFA and solvent B is 0.1% TFA in acetonitrile on a Chromolith Performance RP-18e, 100 × 4.6 mm column (Merck).
Animals and Tumor Growth
Cells (5 × 106) of either the thyroid carcinoma cell line FRO82-2 or the breast carcinoma cell line MCF-7 were suspended in a Matrigel matrix (BD Biosciences) and subcutaneously inoculated into the anterior region of the mouse trunk of female 7- to 8-wk-old BALB/c nu/nu mice (Charles River WIGA). The tumors were allowed to grow for about 10−14 d until approximately 1 cm3 in size. All animals were maintained according to the national animal guidelines.
Organ Distribution with 111In-FROPDOTA
The radiolabeled peptide was injected via the tail vein of the nu/nu mice (0.5–1 MBq per mouse), heterotransplanted with either FRO82-2 thyroid or MCF-7 breast tumors. At 5, 15, 45, and 135 min after injection, the animals were sacrificed. Blood, heart, lung, spleen, liver, kidney, muscle, brain, and the tumor were dissected, blotted dry, and weighed. The radioactivity was measured with a γ-counter (CobraII; Canberra Packard) along with a sample of the injection solution to calculate the percentage injected dose per gram of tissue (%ID/g).
RESULTS
Peptide Synthesis and Labeling
To obtain a DOTA-conjugated analog of FROP-1, the peptide conjugate FROPDOTA was synthesized on a solid support according to the Merrifield strategy. In the identification procedure, the N-terminal was free whereas the C-terminal was attached to the phage scaffold. Therefore, the additional chelate was appended at the C-terminal end of the peptide via an additional lysine. DOTA was chosen as the chelator because it allows one to stably complex several nuclides that complement one another for both diagnostic and therapeutic purposes. 111In is one of the nuclides of choice for in vitro determinations of DOTA conjugates. The acidolytic cleavage and deprotection of the resin-bound FROPDOTA was sluggish because of the slow cleavage rate of the tert-butyl ester groups of DOTA. Therefore, a 2-step procedure was applied: The resin-bound peptide was cleaved with 95:2.5:2.5 TFA:H2O:triisopropylsilane for 1 h and subsequently precipitated with cold ether. To complete the cleavage, the dried precipitate was taken up in neat TFA. After an extended reaction time of 12 h in TFA, a complete conversion was achieved. The conjugate was purified by preparative HPLC and characterized by RP-HPLC and mass spectrometry.
CD
The CD spectra of FROP-1 and FROPDOTA are presented in Figure 1. FROP-1 exhibits a weak rotational strength across the entire spectral range and never crosses from negative to positive. Such CD spectra are typical of peptides with little regular secondary structure, composed principally of coil and several different turn types whose extremes cancel out. As a result, they are difficult to fit with a high coefficient of confidence; in this case, the best fit (0.89) corresponded to 6% helix, 24% coil, and 70% mixed reverse turn. The spectrum of FROPDOTA, on the other hand, is quite different, with a clearly defined positive extreme at ∼194 nm and negative extremes at 207 and 224 nm. Such a spectrum is typical of α-helix, although the red shift of the extremes and their comparatively low amplitude indicate the presence of a significant amount of other structural components. The PEPFIT program fitted this curve with a high coefficient of confidence (0.995), as containing 40% α-helix, 28% coil, and 32% reverse turn.

Structural characteristics of peptides FROP-1 and FROPDOTA: CD spectra of 100 μg/mL FROP-1 (dashed line) and 100 μg/mL FROPDOTA (solid line) in aqueous solutions containing 10% TFE. Mol. Ellip. = molar ellipticity.
Binding Kinetics of 111In-FROPDOTA to Thyroid and Mammary Tumor Cells
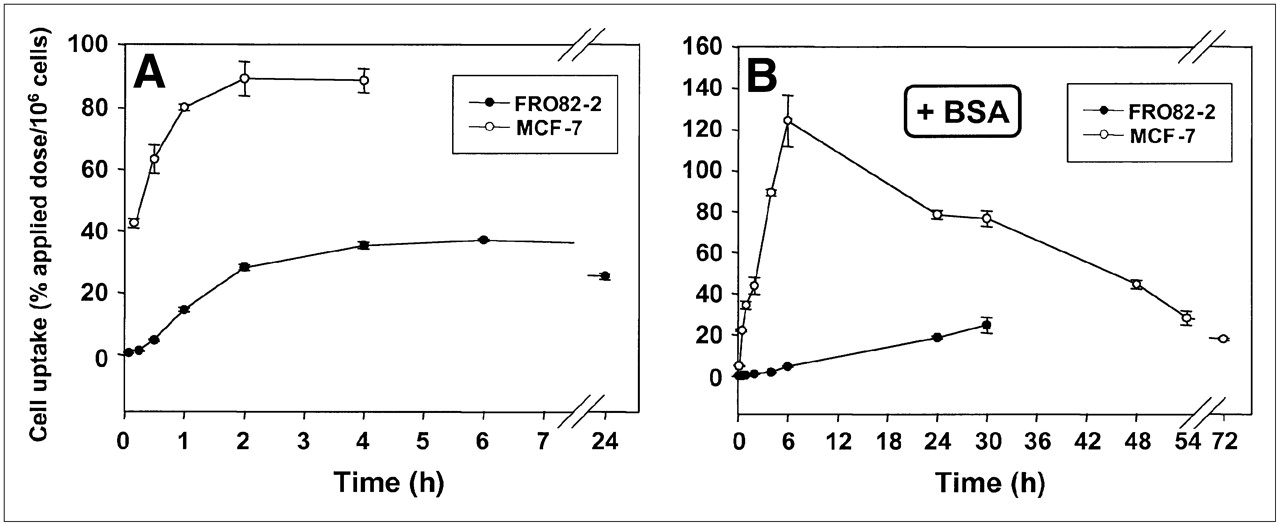
FRO82-2 and MCF-7 cells were seeded into wells and 111In-FROPDOTA was added to the cells and incubated from 5 min up to 24 h (Fig. 2A). The FRO82-2 cells accumulated up to 40% of the applied dose of 111In-FROPDOTA per 106 cells in 4 h, and this level is sustained over the next 24 h with almost no decrease in activity. The MCF-7 cells accumulated up to 90% of the applied dose of 111In-FROPDOTA within 2 h and sustained this level for at least 4 h. A control experiment, which was conducted in parallel under the same conditions with the exception that no cells were seeded into the wells, showed no accumulation of activity over 4 h (data not shown). The addition of 1% BSA to the incubation medium reduced and slowed the accumulation of 111In-FROPDOTA to some extent (Fig. 2B). In the presence of BSA, MCF-7 cells showed an accumulation peak of 120% of the applied dose per 106 cells after 6 h, whereas FRO82-2 cells showed a steady increase of activity over the whole time of the experiment of 30 h, and reached a level of 30% applied dose per 106 cells.
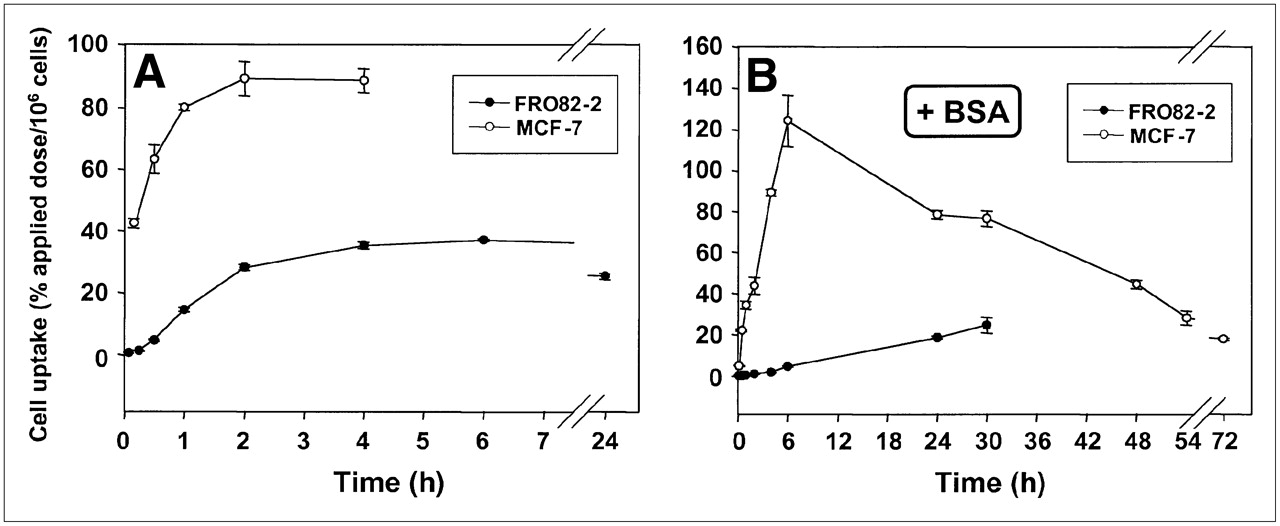
(A and B) In vitro binding kinetics of 111In-FROPDOTA without 1% BSA (A) and with 1% BSA (B).
Receptor Binding
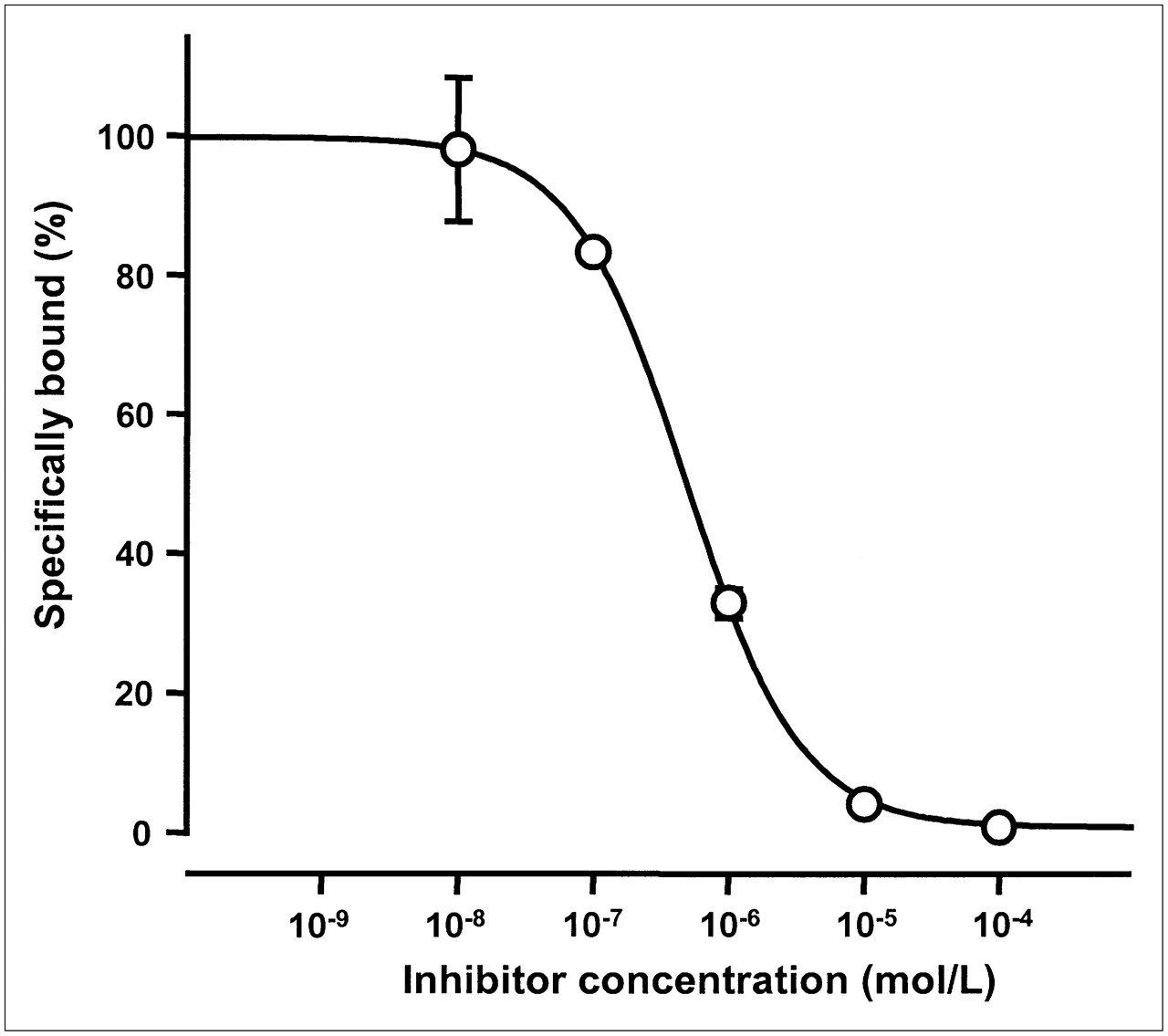
The binding affinity of FROPDOTA was evaluated in a competition binding assay. The ability of FROP-1 to inhibit the binding of 111In-FROPDOTA to FRO82-2 cells at concentrations of 10−4 to 10−8 M was tested after an incubation time of 180 min. As shown in Figure 3, a sigmoidal concentration dependence was observed. The inhibition constant was 494 nM. This inhibition constant in the nanomolar range demonstrates the high specific affinity to the target cells and indicates that the DOTA moiety did not adversely affect the binding affinity of the parent peptide.

In vitro competition assay with FROPDOTA using unlabeled FROP-1 as competitor. Unlabeled FROP-1 in concentrations ranging from 10−4 to 10−8 M was added to cells immediately before 111In-FROPDOTA and incubated for 3 h. All experiments were performed in triplicate; SDs are shown.
Cell Line Binding Profile
Several other cell lines were tested for binding of FROPDOTA, including untransformed primary cells and one nontumor cell line. Besides FRO82-2 cells, the anaplastic thyroid cell line SW1736 showed binding of up to 105% applied dose per 106 cells after a 3-h incubation. Whereas the human breast cancer cell line MCF-7 showed up to 100% applied dose per 106 cells binding, the human colon cell line HCT-116 showed 50% applied dose per 106 cells binding (Fig. 4A). The binding activity could be almost completely blocked by the addition of unlabeled FROP-1 as competitor. Binding of FROPDOTA was not observed in primary umbilical vein endothelial cells (HUVECs). Several human squamous cell carcinoma cell lines have also been tested for binding capacity (Fig. 4B). We found 105% applied dose per 106 cells in HNO 97, 55% applied dose per 106 cells in HNO 210, and 130% applied dose per 106 cells in HNO 223, whereas the human HPV-16 immortalized keratinocyte cell line HPV-16-GM showed only 5% applied dose per 106 cells binding. Again, the binding could be almost completely inhibited by unlabeled FROP-1.
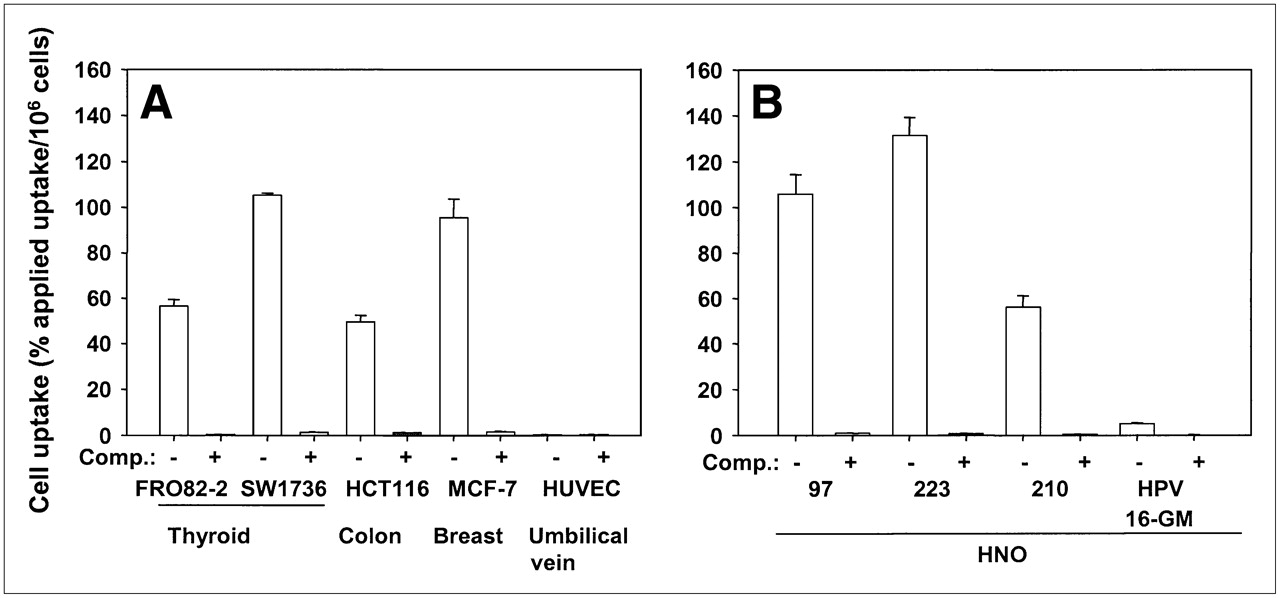
(A and B) In vitro binding studies of FROPDOTA to various tumor cells lines: FRO82-2, SW1736, HCT116, MCF-7, HNO 97, HNO 223, HNO 210, and the nontumor cell line HPV-16-GM and the primary endothelial cells (HUVEC). Cells were incubated with 111In-FROPDOTA without competitor (−) or with 10−4 M unlabeled FROP-1 (+) for 3 h at 37°C. Experiments were performed in triplicate; SDs are shown.
Internalization of FROPDOTA
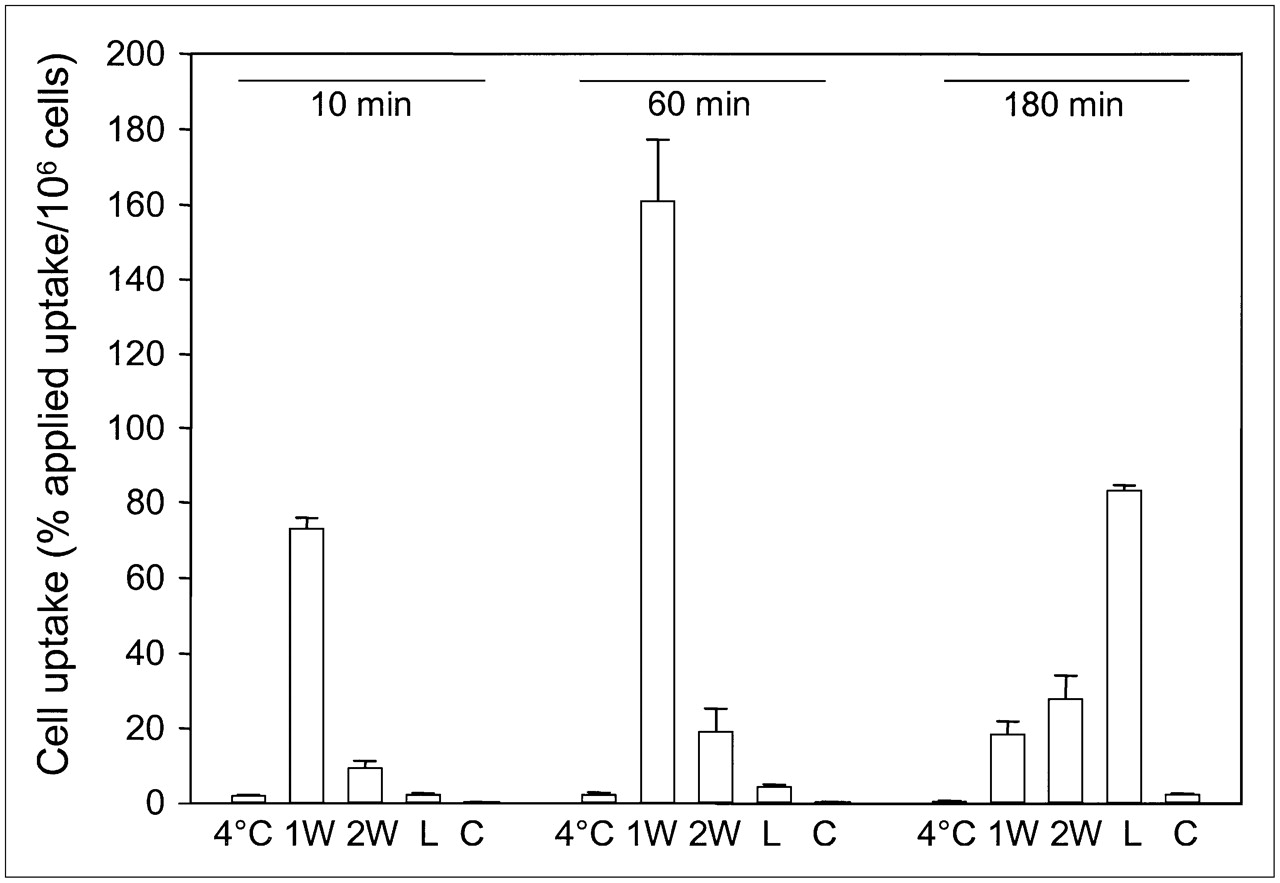
In vitro internalization studies were performed to determine the rate and fraction of internalization of 111In-FROPDOTA in MCF-7 cells (Fig. 5). To distinguish between surface-bound peptide and internalized peptide, 2 acidic wash steps were included in the washing procedure to remove all outside-bound peptide. After 10 min of incubation of the cells with 111In-FROPDOTA at 37°C, about 80% applied dose per 106 cells had already bound to the cells but still remained outside the cells and was removed by the acidic washes. After 1 h of incubation, up to 160% applied dose per 106 cells could be found associated with the cells but most of the activity was removed by the acidic washes, indicating that it was not internalized yet. The fraction of activity recovered after the washes by lysis was increased only from 2% to 5% applied dose per 106 cells. After 180 min of incubation, most of the bound activity was recovered in the internalized fraction, accumulating up to 60% applied dose per 106 cells, whereas only small amounts of activity remained outside the cells. The same experiment conducted at 4°C showed no uptake and no internalization of the tracer. The presence of unlabeled peptide prevented uptake into the cells. The internalization characteristics of 111In-FROPDOTA are shown in Figure 5.
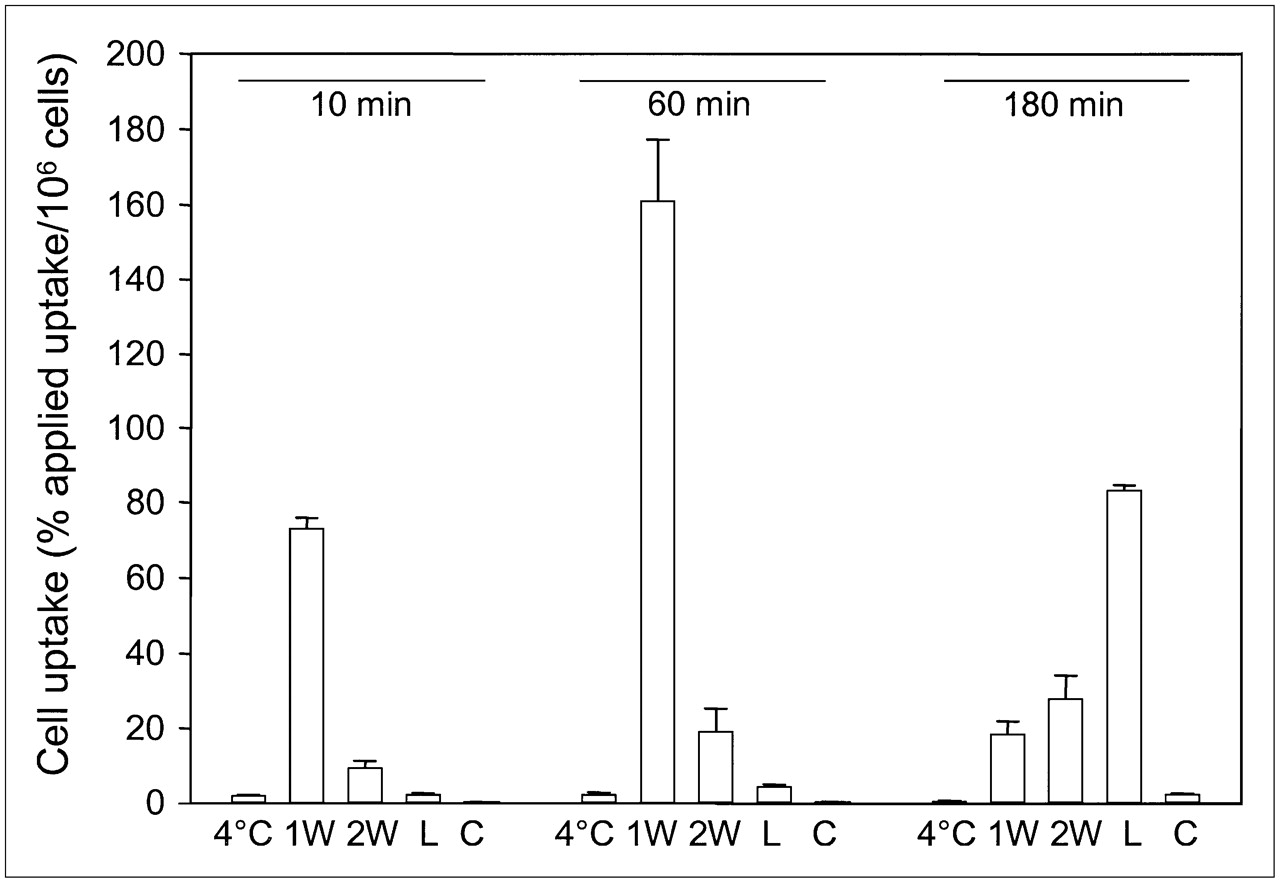
Internalization of 111In-FROPDOTA in MCF-7 cells. Cells were incubated with 3–5 × 105 cpm radioligand for 10 min, 1 h, and 3 h at 37°C. To remove outside-bound peptide, cells were washed with acidic glycine buffer (pH 2.8). Afterward, cells were lysed and internalized radioactivity was measured. 1W = first acidic wash; 2W = second acidic wash; L = lysate; C = control in presence of unlabeled peptide; 4°C = lysate fraction of same experiment conducted at 4°C. Experiments were performed in triplicate; SDs are shown.
Serum Stability of 111In-FROPDOTA
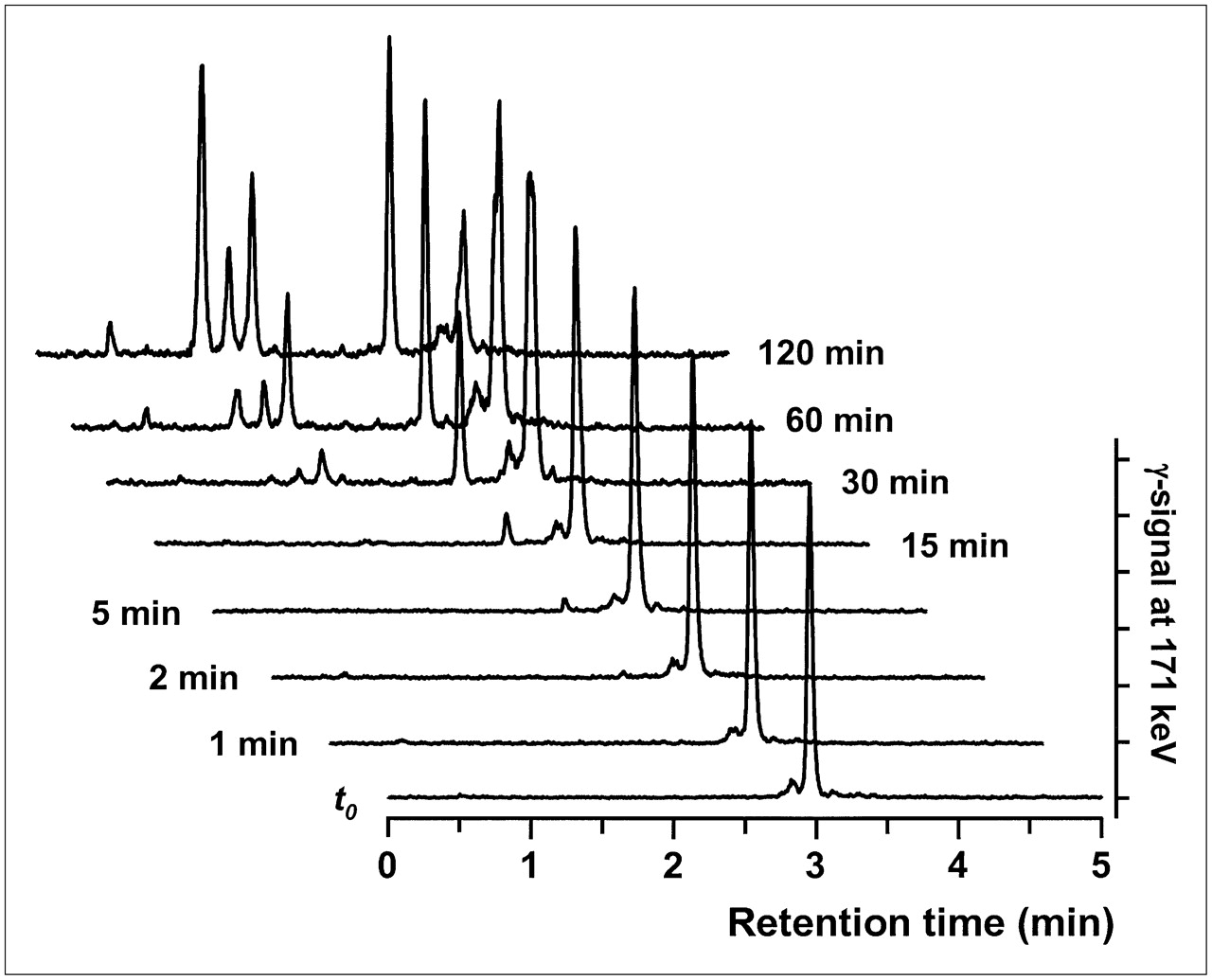
To assess a potential stabilizing effect of the chelate on the FROP-1 peptide, the stability of FROPDOTA was determined in human serum. For these experiments, 111In-FROPDOTA was incubated in human serum at 37°C, aliquots were taken after several incubation intervals, and the peptide was analyzed by HPLC (Fig. 6). After 15 min of incubation, a small second peak appeared with a retention time of approximately 2.5 min, indicating a first degradation product. After 30 min of incubation, a second fragment with a retention time of 1.5 min appeared. Although the fragment peaks increased in size over time and the FROPDOTA peak decreased, full-length peptide was still left after 120 min of incubation. A half-life of 71 min was calculated for the serum stability of FROPDOTA.

RP-HPLC analyses of degradation samples of 111In-FROPDOTA incubated in human serum. t0 = time 0.
Biodistribution of Radiolabeled 111In-FROPDOTA
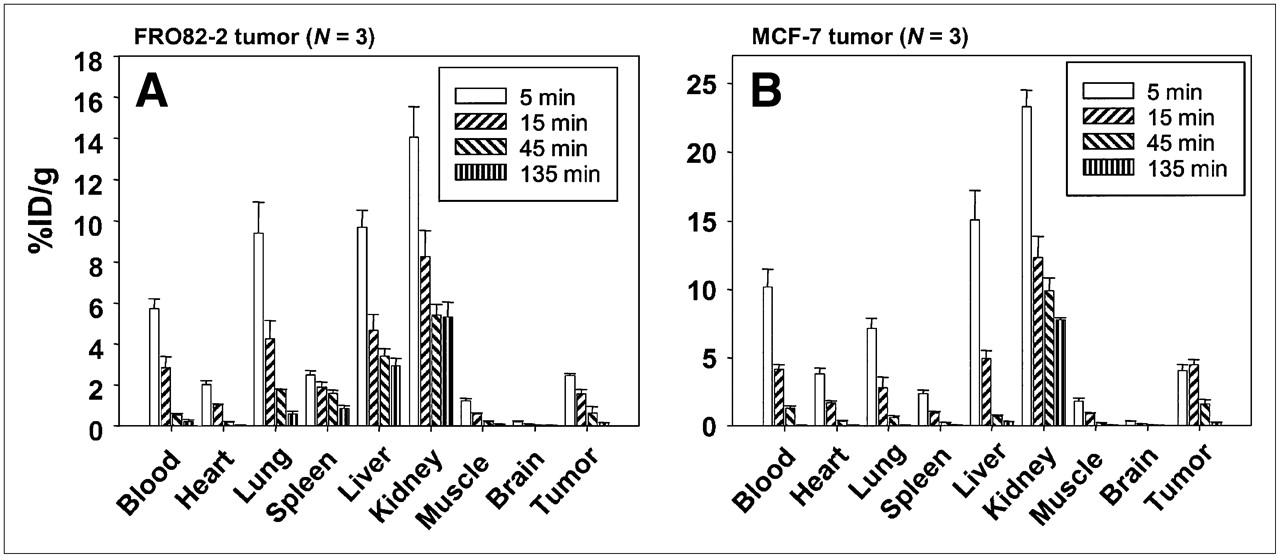
To investigate the distribution of FROPDOTA in vivo, the peptide was labeled with 111In and injected in the tail vein of female nu/nu mice carrying human thyroid tumors (FRO82-2) or human mammary carcinoma (MCF-7) (Fig. 7). The biodistribution in FRO82-2 tumor-carrying mice showed that FROPDOTA reached a level of 2.5 %ID/g in the tumor at 5 min after injection. Thereafter, a steady decrease of radioactivity in the tumor was detected with 1.5 %ID/g after 15 min and 0.6 %ID/g after 45 min. The highest amount of radioactivity was detected in the kidney with almost 14 %ID/g and liver and lung of 10 %ID/g after 5 min, with a rapid decrease of about 6 %ID/g within the following 10 min. The blood showed a level of 6 %ID/g with a decrease to 3 %ID/g after 15 min and almost complete clearance after 45 min. The MCF-7 nude mouse tumor model showed similar kinetics. The tumor accumulated up to almost 5 %ID/g radioactivity at 5 min, and this level remained stable until 15 min after injection before a steep decrease in radioactivity was observed at 45 min after injection with only approximately 1.5 %ID/g in the tumor. Again, the kidneys showed the highest radioactivity with 24 %ID/g after 5 min, decreasing to 12 %ID/g after 15 min and 10 %ID/g after 45 min. Blood radioactivity showed almost 10 %ID/g with a sharp decrease after 15 min to 4.5 %ID/g and almost complete clearance after 45 min. The other organ with high accumulation of radioactivity was the liver, with values up to 15 %ID/g also decreasing fast within 10 min to 5 %ID/g and almost complete clearance after 45 min. Also, all the other organs showed a decrease of radioactivity with time.
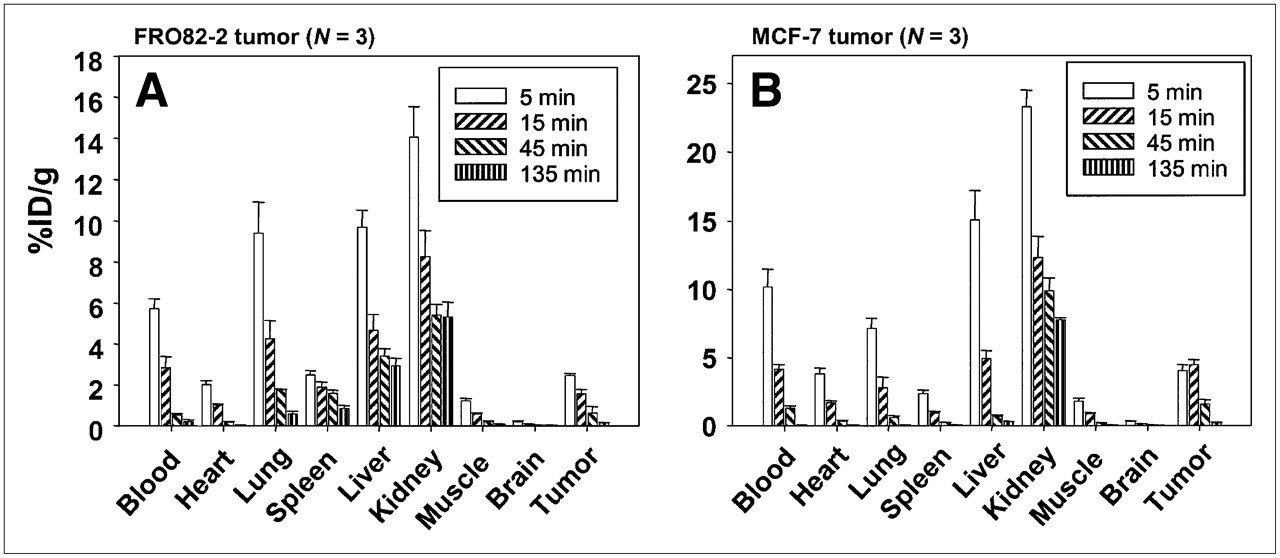
Biodistribution of FROPDOTA in female BALB/c nu/nu mice carrying FRO82-2 tumors (A) (n = 3 animals per time point) or MCF-7 tumors (B) (n = 3 animals per time point). Animals were injected intravenously with 111In-FROPDOTA, and the radioactivity was measured in tumor and control organs after 5, 15, 45, and 135 min; SDs are shown.
DISCUSSION
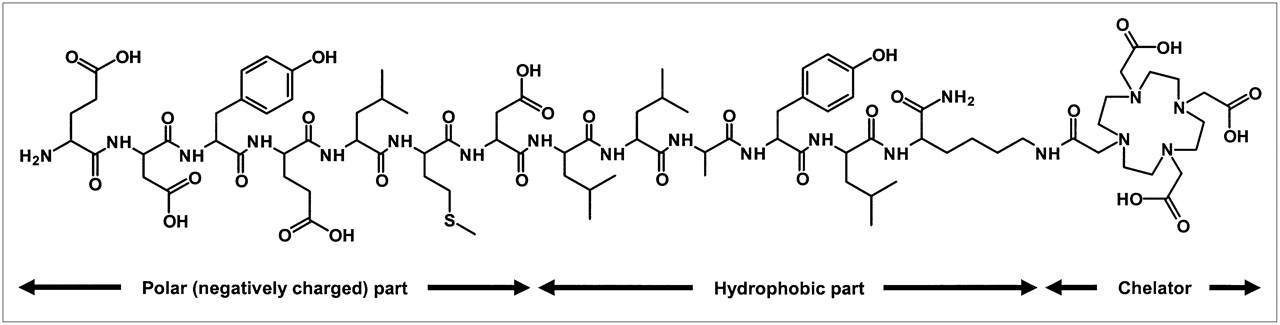
The peptide FROP-1 was discovered recently by biopanning of a peptide phage-display library on the thyroid carcinoma cell line FRO82-2 (8). This “fresh-from-the-cradle” peptide revealed interesting in vitro and in vivo features (8). To modify the secondary structure and to improve the stability of the label for in vivo applications, a chelator has now been attached to the C-terminus of the peptide and used to label the peptide with 111In. A CD analysis was performed to allow a comparison between the original FROP-1 peptide and FROPDOTA, the peptide with the attached chelator. FROPDOTA was remarkably more structured in solution than FROP-1, which essentially has no regular secondary structure components and is a tangle of coil and reverse turns. This structure is typical of that adopted by chemically denatured proteins. It is interesting that, although the fraction of α-helix in FROPDOTA is increased considerably, the coil level is scarcely altered. The increased helix content appears to occur at the cost of turn, with the coil component remaining unchanged. This suggests that the peptide may tend to adopt the helical conformation on association or complexation with other proteins. Such pronounced secondary structure is not typical for peptides of this size. As shown in Figure 8, the peptide sequence of FROPDOTA is of amphiphilic character, a fact that might explain the structural features.
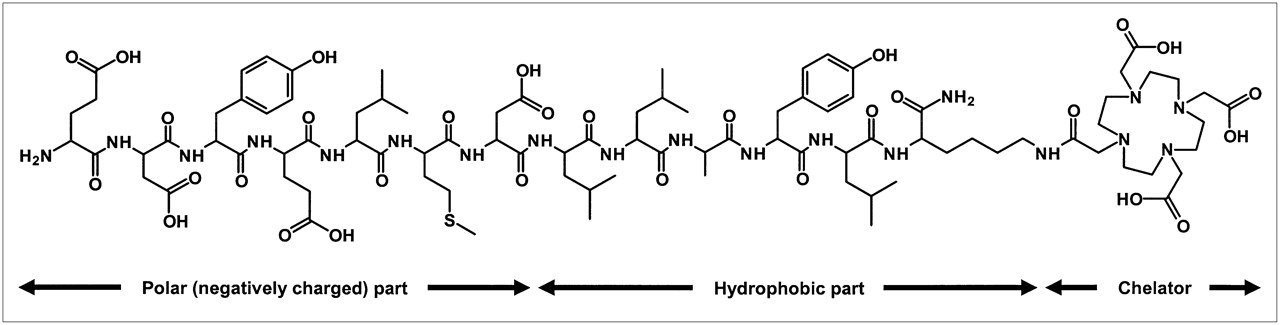
Chemical structure of FROPDOTA (H-EDYELMDLLAYLK(DOTA)-NH2). Amino acids 1–7 comprise 4 carboxylic acids. The strong negative net charged part of molecule is linked to the chelator via a hydrophobic molecule part.
The differences observed between the iodine-labeled FROP-1 and the indium-chelator–labeled peptide FROPDOTA in the in vitro test system are considerable.
The conjugation of the DOTA moiety increases the affinity by a factor of approximately 20: The binding affinity of 111In-FROPDOTA to FRO82-2 cells is 494 nM; the affinity of 125I-FROP-1 for FRO82-2 is approximately 8 μM.
The peptide 125I-FROP-1 showed binding to MCF-7 cells of up to 6% applied dose per 106 cells, whereas 111In-FROPDOTA reached 100% applied dose per 106 cells. These high values meant that during the time course of the experiment the activity actually “drained” out of the medium and associated with the tumor cells. The same effect was observed with the FRO82-2 cells, as well as the SW1736, HCT116, and HNO 97, HNO 210, and HNO 223 cells—all showing very strong binding of FROPDOTA. In contrast, no binding was observed either for FROPDOTA or for FROP-1 for the negative control cell line HPV-16GM and the primary endothelial cells (HUVEC).
The peptide FROPDOTA showed slower binding kinetics in vitro compared with FROP-1. The maximum binding for FROP-1 was around 10–20 min, whereas FROPDOTA reached its maximum at 2–4 h, remaining unchanged for almost 24 h. The fixed α-helix structure of FROPDOTA apparently induces a strong change in secondary structure and constitutes an avid binding conformation of the peptide (14). In contrast, FROP-1 has a more flexible structure.
A difference in the serum stability was observed between FROPDOTA and the FROP-1 peptide. This would be expected because of the strong changes of the secondary structure. The N-terminal glutamate of FROP-1 is cleaved off almost immediately in the serum. For FROPDOTA, this is not the case, as the fragments that appear seem to be different when one considers the retention time.
When bound to their cellular target, carrier peptide conjugates dissociate dependent on their binding affinity. In most cases, the affinity of peptide protein interactions is too weak to ensure a stable cell binding for the whole period of the therapeutic effect. Therefore, the internalization is a prerequisite for the treatment with carrier peptide conjugates, in general, and we believe this property should be examined in the preclinical investigation of carrier peptides. 111In-FROPDOTA revealed a high rate of internalization but the internalization kinetics was slow.
The in vivo study showed a faster blood clearance of FROPDOTA in comparison with FROP-1. Although the level of FROP-1 in the blood of MCF-7 tumor-bearing mice reached only 5.4 %ID/g at 5 min after injection, it decreased to only 4.1 %ID/g after 15 min and to 3.2 %ID/g after 45 min. FROPDOTA in blood of MCF-7 tumor-bearing mice showed an activity level of 10.2 %ID/g after 5 min, which decreased to 4.1 %ID/g after 15 min and 1.3 %ID/g after 45 min. This rapid blood clearance could be explained by the higher polarity of the DOTA conjugate. The in vitro data suggested a maximum binding at 2–4 h but, in the in vivo situation, rapid clearance from the blood does not allow the peptide to reach its maximum binding capacity. This could also be reflected in the different binding kinetics of the 2 cells lines. The MCF-7 cells lines showed faster binding kinetics in vitro as compared with the FRO82-2; in vivo the MCF-7 tumors showed more activity in the tumor than the FRO82-2, which could be the result of the faster peptide binding of the MCF-7 tumor cells. The fast renal clearance is caused by the hydrophilic chelator DOTA (15). Consequently, derivatives with a prolonged circulation time to provide the time necessary for successful interaction with the target cells in vivo must be investigated as a next step (16).
CONCLUSION
We were able to show that the tumor-targeting properties of FROP can be modulated to increase tremendously the binding affinity of the peptide used without the phage scaffold. This effect can be attributed to structural changes observed due to the conformational changes induced by the chelator attached at the C-terminus of the peptide. Our findings highlight the potential of peptides identified by display techniques when carefully probing the pharmacologic properties of stabilized or conformationally restrained peptide derivatives.
Acknowledgments
The authors thank Ulrike Hebling, Uschi Schierbaum, Sigrid Peschke, and Karin Leotta for excellent technical assistance. Special thanks are given to Kerstin Schmidt for supplying the HUVEC cells, Christel Herold-Mende for providing the HNO cell lines, and Pascal Tomakidi for the HPV-16-GM cell line. This study was supported by grants from the Deutsche Forschungsgemeinschaft (grants HA2901/2-1 and HA2901/2-2).
Footnotes
-
COPYRIGHT © 2007 by the Society of Nuclear Medicine, Inc.
References
- Received for publication September 25, 2006.
- Accepted for publication May 31, 2007.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}