


Abstract
Small-molecule ligands specific for prostate-specific membrane antigen (PSMA) have the potential to improve prostate cancer imaging. However, highly charged ligands are difficult to label with 99mTc and to purify. In this study, we present an adamantane-trimerized small molecule that has nanomolar binding to PSMA and also has 12 negative charges. Methods: To convert this molecule into a clinically viable SPECT diagnostic, we have developed a simple, cartridge-based, solid-phase prelabeling strategy that, within 25 min, converts readily available and inexpensive 99mTc-pertechnetate into a chemically pure complex, with a reactive N-hydroxysuccinimide (NHS) ester, in neat organic solvent. This stable intermediate can label any amine-containing small molecule or peptide with 99mTc in 1 step, with high specific activity and without the need for high-performance liquid chromatography (HPLC). Results: Solid-phase conversion of 99mTc-pertechnetate to 99mTc-MAS3-NHS (MAS3 is S-acetylmercaptoacetyltriserine) could be completed in 25 min, with >99% radiochemical purity and with no coligands present. This intermediate was then conjugated to adamantane-trimerized GPI (2[(3-amino-3-carboxypropyl)(hydroxy)(phosphinyl)-methyl]pentane-1,5-dioic acid) in 1 step with >95% yield and no need for HPLC purification. The final molecule bound specifically to living human tumor cells expressing PSMA on their surface. Quantitative comparison was made among GPI monomer, GPI trimer, and their 99mTc-derivatives. Conclusion: Our study describes a simple cartridge-based conversion of 99mTc-pertechnetate to a useful, preloaded NHS ester intermediate that takes only 25 min to prepare and results in >99% radiochemical purity. Using this chemistry, we produced a high-specific-activity, 99mTc-labeled, PSMA-targeted small molecule and demonstrate γ-ray radioscintigraphic imaging of living human prostate cancer cells.
- 99mTc-pertechnetate
- S-acetylmercaptoacetyltriserine (MAS3)
- solid-phase chemistry
- SPECT
- prostate-specific membrane antigen
Prostate cancer is diagnosed in approximately 230,000 men each year in the United States, and >30,000 of these will die of the disease (1). Every patient with biopsy-confirmed cancer requires clinical and radiographic staging to determine whether cancer has spread outside the capsule or has metastasized to regional lymph nodes or to bone. For patients already diagnosed with metastases, nuclear imaging could be used to pinpoint sites of disease and to quantify response to therapy.
SPECT is a nuclear medicine technique by which γ-ray emissions from a radioisotope are used to localize cancer and its products. The major radioisotope used clinically is 99mTc, which emits a monoenergetic, 140-keV photon, has a 6-h half-life, and has a low absorbed dose. Most important, it is readily available at most institutions and is extremely inexpensive to produce, typically $10.00 for 740 MBq (20 mCi). The major problem with 99mTc is that it is eluted using saline as sodium pertechnetate (Na99mTcO4) in the +VII oxidation state, which requires reduction to the +V oxidation state, as well as exchange chelation, for stability and subsequent chemical manipulation. Any chemistry capable of converting sodium pertechnetate to a more useful form would have immediate impact on diagnostic agent development.
Given its half-life, 99mTc is an ideal radiotracer for any targeting molecule having a total-body clearance of ≤6 h, such as small molecules, peptides, and single-chain antibodies. Of these, small molecules have distinct advantages for tumor targeting due to rapid biodistribution, rapid clearance, and excellent penetration into solid masses. Our group (2) and others (3,4) have reported small molecules with high-affinity binding to prostate-specific membrane antigen (PSMA), a type II transmembrane glycoprotein expressed at high levels on normal and malignant prostate epithelial cells (5,6). Our GPI (2[(3-amino-3-carboxypropyl)(hydroxy)(phosphinyl)-methyl]pentane-1,5-dioic acid) molecule is a 311-Da monomeric small molecule with an affinity of 9 nM and a net charge of −4 after conjugation to contrast agents or radiotracers.
However, 2 major problems impede the use of GPI and similar molecules in diagnostic imaging. First, in its monomeric form, the phosphate anion in serum competes with it for binding to PSMA. Second, its chemical structure resembles classic exchange ligands, such as tartaric acid, which are required to stabilize 99mTc during its reduction. Hence, 99mTc could be chelated by GPI itself and block binding to PSMA, and HPLC purification of the final product using conventional labeling strategies is extremely difficult (7). In this study, we solve both problems by developing a simple and rapid cartridge-based, solid-phase prelabeling strategy for 99mTc and also describe an adamantane-based trimeric form of GPI that retains full binding to living prostate cancer cells in the presence of serum.
MATERIALS AND METHODS
Reagents
MGI Pharma compound 11245-36 (GPI) was synthesized as described previously (8). Ultradry dimethyl sulfoxide (DMSO) was purchased from Acros Organics. HPLC-grade triethylammnonium acetate, pH 7 (TEAA), was from Glen Research. HPLC-grade water was from American Bioanalytic. Triserine was from Bachem. N-succinimidyl-S-acetylthioacetate (SATA) was from Pierce. N,N,N′,N′-Tetramethyl-O-(N-succinimidyl)uronium tetrafluoroborate (TSTU) was from Advanced Chemtech. All other chemicals were purchased from Fisher Scientific and were American Chemical Society (ACS)- or HPLC-grade.
HPLC/Mass Spectrometry Platform
The HPLC/mass spectrometry platform used for purification of both nonradioactive and radioactive tumor-targeting small molecules and peptides has been described in detail previously (7). Briefly, the system is composed of a Waters model 1525 binary pump, a model 2487 ultraviolet detector (Waters), a Sedex model 75 (Richards Scientific) evaporative light scatter detector (ELSD) with the nebulizer modified to reduce band broadening at low flow rates, a model FC-3200 high-sensitivity photomultiplier tube γ-detector (Bioscan), and a Waters fraction collector, all housed within a Capintec hot cell equipped with a model CRC-15R (Capintec) dose calibrator. For nonradioactive reactions, column eluate was split into a Waters LCT electrospray time-of-flight (ES-TOF) mass spectrometer.
Preparative Synthesis of S-Acetylmercaptoacetyltriserine (MAS3)
Ten milligrams (36 μmol) of triserine were dissolved in 350 μL of water. One equivalent (5 μL, 36 μmol) of the base triethylamine (Et3N) was added, followed by 2 equivalents (16.6 mg, 72 μmol dissolved in 160 μL N,N-dimethylformamide [DMF]) of SATA. The reaction mixture was vortexed at room temperature for 3 h. An additional equivalent (8.3 mg, 36 μmol dissolved in 80 μL DMF) of SATA was then added, and vortexing was continued for an additional 2 h.
To confirm completion of the reaction, a 10-μL sample was analyzed by reverse-phase HPLC (RP-HPLC) using a 4.6 × 150 mm Symmetry (Waters) C18 column and a linear gradient from 0% to 15% B over 35 min, starting 2 min after injection, at 1 mL/min, where A = H2O + 0.1% formic acid and B = acetonitrile + 0.1% formic acid. MAS3 eluted at a retention time (Rt) = 11.6 min as detected by the ELSD, with its mass confirmed by ES-TOF mass spectrometry. Preparative purification was performed on an HPLC system described in detail previously (7) and equipped with a 5-mL sample loop, after dilution into a final volume of 5 mL of H2O + 0.1% trifluoroacetic acid (TFA), and using a 19 × 150 mm Symmetry C18 column. The gradient consisted of 0% B for 3.5 min, then 0%–15% B over 35 min at 7 mL/min, where A = H2O + 0.1% TFA and B = acetonitrile + 0.1% TFA. MAS3 eluted at Rt = 21.8 min using ELSD detection. Fractions containing product were pooled and lyophilized. MAS3 was obtained as a white powder in 57% isolated yield (8.0 mg, 20.5 μmol), with the expected mass confirmed by ES-TOF mass spectrometry, and a purity of ≥98%.
Solid-Phase Labeling of MAS3 with 99mTc-Pertechnetate
One-hundred fifty microliters of a 50% slurry of Chelex 100 resin (Bio-Rad) in 50 mM 2-(N-morpholino)ethanesulfonic acid (MES) buffer, pH 5.0, were added to an empty micro Bio-Spin (Bio-Rad) chromatography column, washed once with MES buffer, and centrifuged at 3,000g for 10 s; 1.2 mg (3 μmol) of MAS3 were dissolved in 1 mL of water. One milligram (4 μmol) of stannous (II) chloride dihydrate was dissolved in 1 mL of 10 mM HCl. Then 100 μL of MAS3 and 35 μL of stannous chloride were mixed well and added to the Chelex resin. 99mTc-pertechnetate (185–370 MBq [5–10 mCi]) in 100 μL of saline, eluted directly from a 99Mo generator with saline, were added to the tube. The tube was capped, wrapped in parafilm, and boiled for 10 min in a water bath. 99mTc loading of MAS3 was monitored by RP-HPLC using a 4.6 × 75 mm Symmetry C18 column with a linear gradient from 0% to 60% B over 30 min, starting 2 min after injection, at 1 mL/min, where A = 10 mM TEAA and B = absolute MeOH. 99mTc-MAS3 eluted at Rt = 14.1 min. In separate experiments, the limits of this reaction were determined to be 370 MBq (10 mCi) 99mTc-pertechnetate and 1.3 μmol of MAS3 using 150 μL of 50% Chelex 100 slurry.
A 10-mg Oasis HLB cartridge (catalog no. 186000383) was activated using methanol, then H2O, and equilibrated with H2O, pH 4.0 (pH adjusted with 10 mM HCl). The 99mTc-MAS3 solution was diluted with 1 mL of H2O, pH 4.0, and bound to the cartridge. The cartridge was washed with 10 mL of H2O, pH 4.0, to remove 99mTc-pertechnetate, tin, and free MAS3; then it was purged completely with nitrogen. Finally, 99mTc-MAS3 was eluted with 400 μL dry DMSO or DMF.
Synthesis of [99mTc-MAS3]-NHS
After 200 μg (0.7 μmol) of TSTU powder were placed in a 1.5-mL Eppendorf tube, 185–370 MBq (5–10 mCi) of purified 99mTc-MAS3 in 400 μL DMSO or DMF and 0.5 μL (2.9 μmol) of neat (5.74 M) N,N-diisopropylethylamine (DIEA) were added. The top was sealed and the solution was incubated at 60°C for 10 min. The reaction was diluted to 1 mL final volume by the addition of 600 μL of dichloromethane:hexane (6:4). The reactants and by-products were removed by passage through Waters Oasis MCX (catalog no. 186000252) and MAX (catalog no. 186000366) cartridges attached in tandem. The flow-through, containing pure [99mTc-MAS3]-NHS ester, was concentrated by loading on an Oasis HLB cartridge, washing with of 1 mL of dichloromethane:hexane (6:4), and eluting using 500 μL of either DMF or DMSO. The purity of the compound was assessed by RP-HPLC using a 4.6 × 75 mm Symmetry C18 column with a linear gradient from 0% to 60% B over 30 min, starting 2 min after injection, at 1 mL/min, where A = 10 mM TEAA and B = absolute MeOH. [99mTc-MAS3]-NHS eluted at Rt = 23.5 min. The specific activity of [99mTc-MAS3]-NHS was estimated by conjugation to tryptophan and measurement of 280-nm absorbance of the product using the same RP-HPLC conditions as those used to assess purity.
Synthesis and Purification of Radiolabeled PSMA Ligands
Covalent conjugation of GPI derivatives with [99mTc-MAS3]-NHS was performed by the addition of 100 μL (10 μmol) of 100 mM triethylamine in dry DMSO to 10 μL (0.1 μmol) of a 10 mM solution of GPI derivative in a total of 400 μL dry DMF/DMSO, followed by the addition of 200 μL of [99mTc-MAS3]-NHS (185–259 MBq [5–7 mCi]) in dry DMSO. Constant stirring at room temperature was maintained for 40–90 min, until the reaction was completed. The radiolabeled ligands were analyzed by RP-HPLC on a 4.6 × 75 mm Symmetry C18 column using a linear gradient from 0% to 60% B over 25 min, beginning 2 min after injection, at a flow rate of 1 mL/min, where A = 10 mM TEAA, pH 7.0, and B = absolute MeOH.
Synthesis of Re-MAS3
A procedure reported previously (9) was used with slight modification. MAS3 (4.8 mg, 16 μmol) was dissolved in 1.5 mL of water. Stannous chloride dihydrate (5.4 mg, 24 μmol) in 1.5 mL of 0.1 M citrate buffer, pH 5.0, and NaReO4 (6.6 mg, 24 μmol) in 1.5 mL of H2O were added to the MAS3 solution. The reaction mixture was stirred at 90°C for 1 h. After cooling to room temperature, Re-MAS3 was purified on an Oasis HLB cartridge and eluted with DMSO. The purity was assessed by liquid chromatography/mass spectrometry (LC/MS) on a 4.6 × 150 mm Symmetry C18 column using a linear gradient from 0% to 50% B over 15 min, beginning 2 min after injection, at a flow rate of 1 mL/min, where A = H2O + 0.1% formic acid and B = acetonitrile + 0.1% formic acid. Retention time was 6.4 min.
Synthesis of [Re-MAS3]-NHS
Fifty microliters (3 μmol) of a 60 mM TSTU solution in DMSO and 200 μL (2 μmol) of a 10 mM Re-MAS3 solution in DMSO were mixed together; then 25 μL (5 μmol) of 200 mM N,N-diisopropylethylamine and 25 μL of DMSO were added. The solution was vortexed at room temperature for 40 min. After dilution with dichloromethane, the NHS ester was purified using Oasis MCX, MAX, and HLB cartridges connected in series as described earlier. The purity was assessed by LC/MS on a 4.6 × 150 mm Symmetry C18 column using a linear gradient from 0% to 50% B over 15 min, beginning 2 min after injection, at a flow rate of 1 mL/min, where A = H2O + 0.1% formic acid and B = acetonitrile + 0.1% formic acid. Retention time was 10.4 min.
Synthesis and Purification of [Re-MAS3]-Conjugates
Covalent conjugation of GPI derivatives with [Re-MAS3]-NHS was performed by the addition of 0.1 mL (10 μmol) of 100 mM triethylamine in dry DMSO to 0.1 mL (1 μmol) of a 10 mM solution of GPI derivatives in dry DMSO or DMF, followed by addition of 0.2 mL (2 μmol) of a 10 mM [Re-MAS3]-NHS solution in dry DMSO. Constant stirring at room temperature was maintained for 2 h. The conjugates were purified by preparative HPLC (Symmetry C18 column, 19 × 150 mm, 7 μm) using a linear gradient from 0% to 50% B over 27 min, starting 5 min after injection, with a flow rate of 10 mL/min. The collected fractions were lyophilized, and purity was assessed by LC/MS on a 4.6 × 150 mm Symmetry C18 column using a linear gradient from 0% to 50% B over 15 min, beginning 2 min after injection, at a flow rate of 1 mL/min, where A = H2O + 0.1% formic acid and B = acetonitrile + 0.1% formic acid.
Quantification of Serum Stability
The stabilities of [99mTc-MAS3]-GPI compounds were tested by incubation in the absence (phosphate-buffered saline [PBS] only) or presence of 100% calf serum for 4 h at 37°C. Stability and transmetallation were quantified using high-resolution chromatography. For PBS experiments, a 4.6 × 75 Symmetry C18 column was used as described earlier for radiolabeled PSMA ligands. For serum experiments, an 8 × 300 mm, 200-Å Diol (YMC, catalog no. DL20S053008WT) gel-filtration column was used with PBS as the mobile phase. Gel-filtration molecular weight (M.W.) markers (Bio-Rad) were: M1 = thyroglobulin (670 kDa), M2 = γ-globulin (158 kDa), M3 = ovalbumin (44 kDa), M4 = myoglobin (17 kDa), and M5 = vitamin B12 (1.3 kDa).
High-Throughput, Radioactive Live Cell Binding and Affinity Assay
Human prostate cancer cell lines, PSMA-positive LNCaP and PSMA-negative PC-3, were obtained from the American Type Culture Collection. The PSMA-negative human bladder cancer cell line TsuPR1 was a generous gift of Dr. John T. Isaacs (Johns Hopkins University, Baltimore, MD). Cell lines were cultured at 37°C under humidified 5% CO2 in RPMI 1640 medium (Mediatech Cellgro) supplemented with 10% fetal bovine serum (Gemini Bio-Products) and 5% penicillin/streptomycin (Cambrex Bioscience). Cells were split onto 96-well filter plates (model MSHAS4510; Millipore) and grown to 50% confluence (approximately 35,000 cells per well) over 48 h.
To assign absolute affinity to each compound, a homologous competition assay was used with the 99mTc-labeled version as the tracer and the relabeled version as the test compound. To avoid internalization of the radioligand due to constitutive endocytosis (2), live cell binding was performed at 4°C. Cells were washed 2 times with ice-cold Tris-buffered saline (TBS), pH 7.4, and incubated for 20 min at 4°C with 0.02 MBq (0.5 μCi) of radiotracer in the presence or absence of the test compound. Cells were then washed 3 times with TBS using a Millipore vacuum manifold (catalog no. MSVMHTS00), and the well contents was transferred directly to 12 × 75 mm plastic tubes placed in γ-counter racks. Transfer was accomplished using a modified (Microvideo Instruments) 96-well puncher (MAMP09608; Millipore) and disposable punch tips (MADP19650; Millipore). Well contents were counted on a model 1470 Wallac Wizard (Perkin Elmer) 10-detector γ-counter, and curves were fit using Prism version 4.0a (GraphPad) software.
Near-Infrared (NIR) Fluorescence and γ-Radioscintigraphic Imaging
To assess viability and to verify confluence, living cells were loaded with the NIR fluorophore IR-786 by adding it to the cell culture medium at 1 μM for 30 min at 37°C before the start of the experiment (10). Cells were incubated as described earlier with 3.7 MBq (100 μCi) of 99mTc-labeled compound per well, in the buffer being tested, for 20 min at 4°C, before extensive washing of the filter plate. Simultaneous white light and NIR fluorescence imaging of plates was performed as described in detail previously (11,12). γ-Radioscintigraphy was performed with an Isocam Technologies Research Digital Camera equipped with a ½-in. NaI crystal, 86 photomultiplier tubes, and a high-resolution, low-energy lead collimator.
RESULTS
High-Affinity, Multimeric Small Molecules for Targeting PSMA
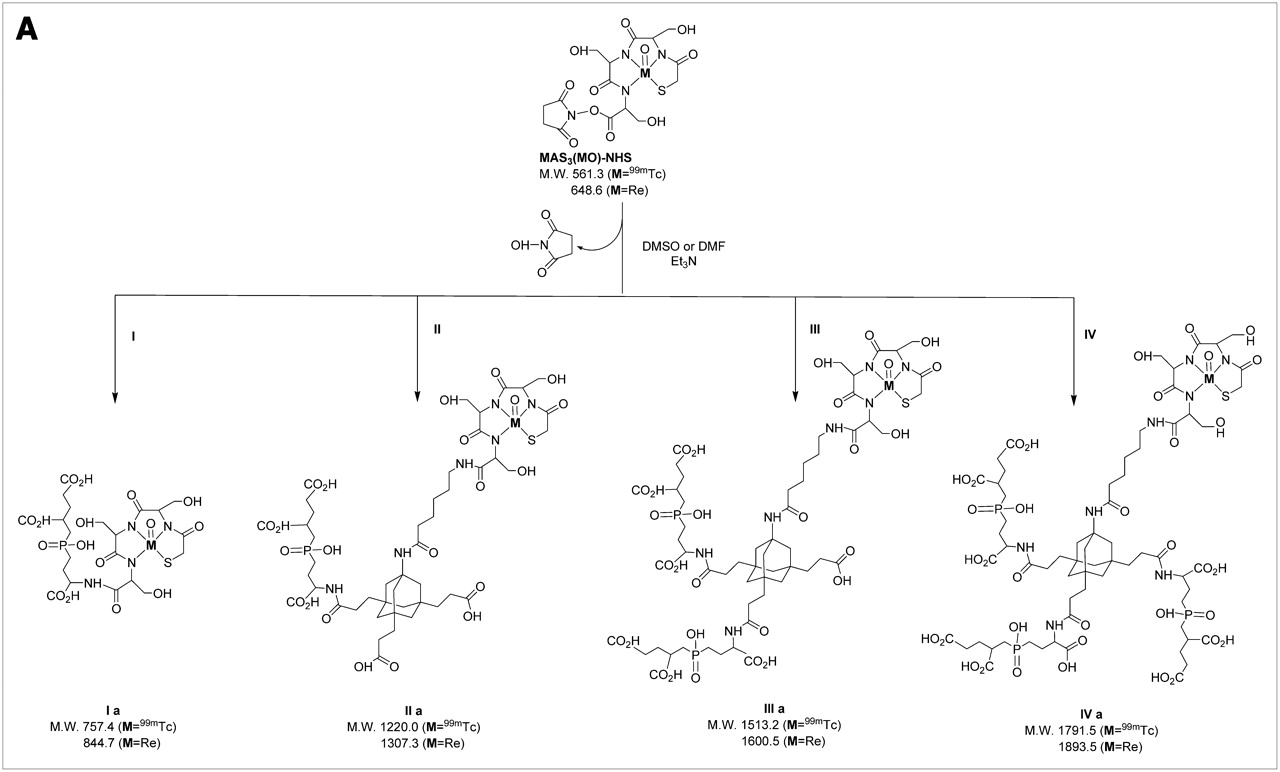
Our group has previously developed a high-affinity (9 nM), monomeric small molecule (311 Da) termed GPI, which is engineered to bind the active site of PSMA (2). Unfortunately, inorganic anions in serum—such as phosphate—compete with monomeric GPI, rendering it marginal for use as an in vivo diagnostic (2). The work of Whitesides and colleagues (13) suggests that by decreasing the off-rate, multivalency can permit a small molecule to “out-compete” endogenous competitors. To test this hypothesis, while maintaining overall small size, we have developed a tri-NHS ester derivative of adamantane (14), which permits conjugation of up to 3 targeting ligands (such as GPI) in addition to a contrast agent or radiotracer. Recently, we have added a 6-carbon linker to improve radiotracer conjugation and have purified a series of GPI derivatives with increasing valency (Fig. 1). As shown in Table 1, conversion of monomeric GPI (I) to an adamantane trimer (IV) results in an affinity improvement of over 1.5 logarithms and retention of affinity in 100% serum.
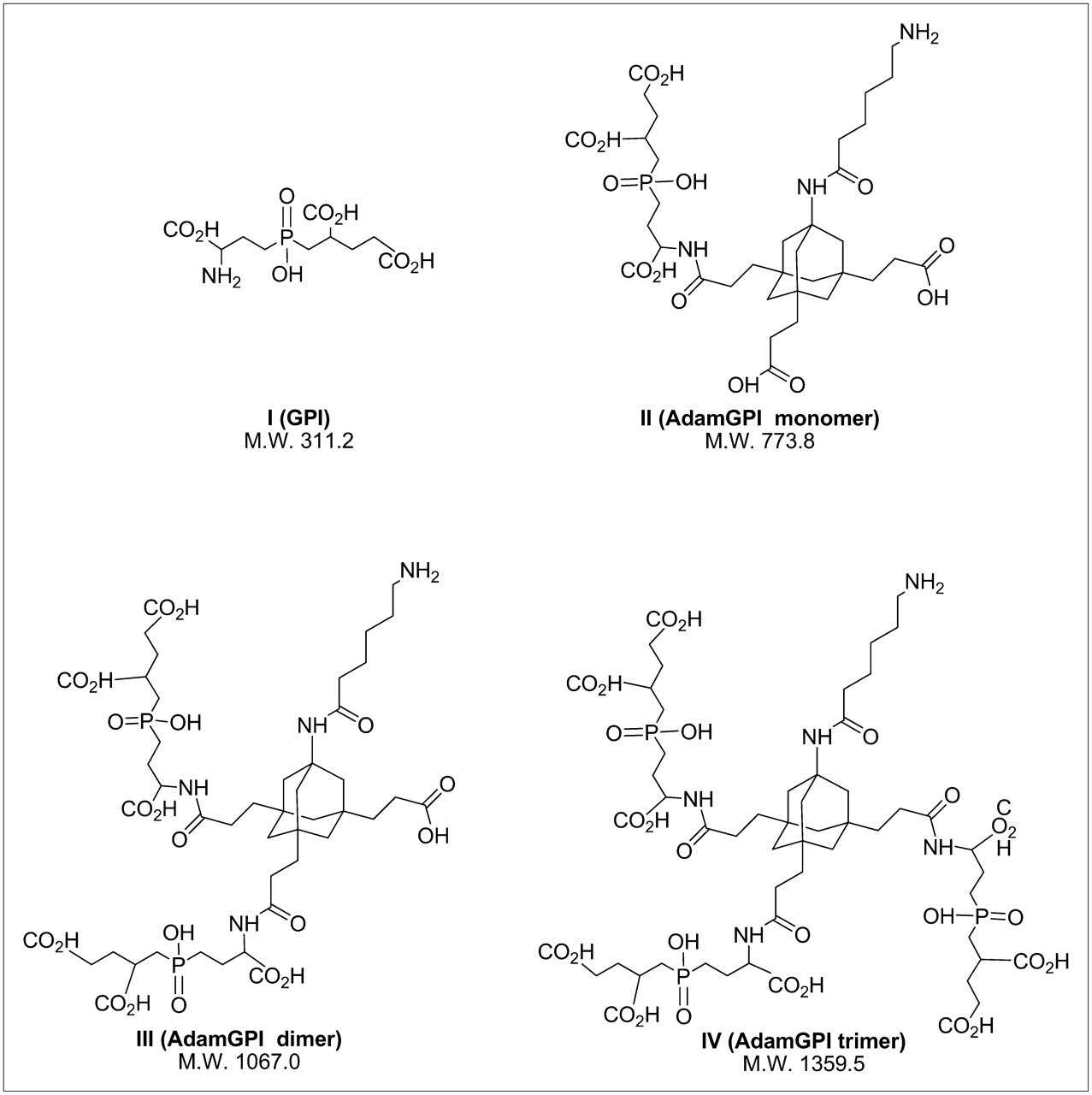
Highly anionic PSMA-targeting small-molecule ligands: chemical structures and M.W. for PSMA-targeting ligands used in this study.
Live Cell-Binding Affinity of Small Molecules Targeted to PSMA
Solid-Phase Prelabeling for Preparation of 99mTc-MAS3 Conjugates
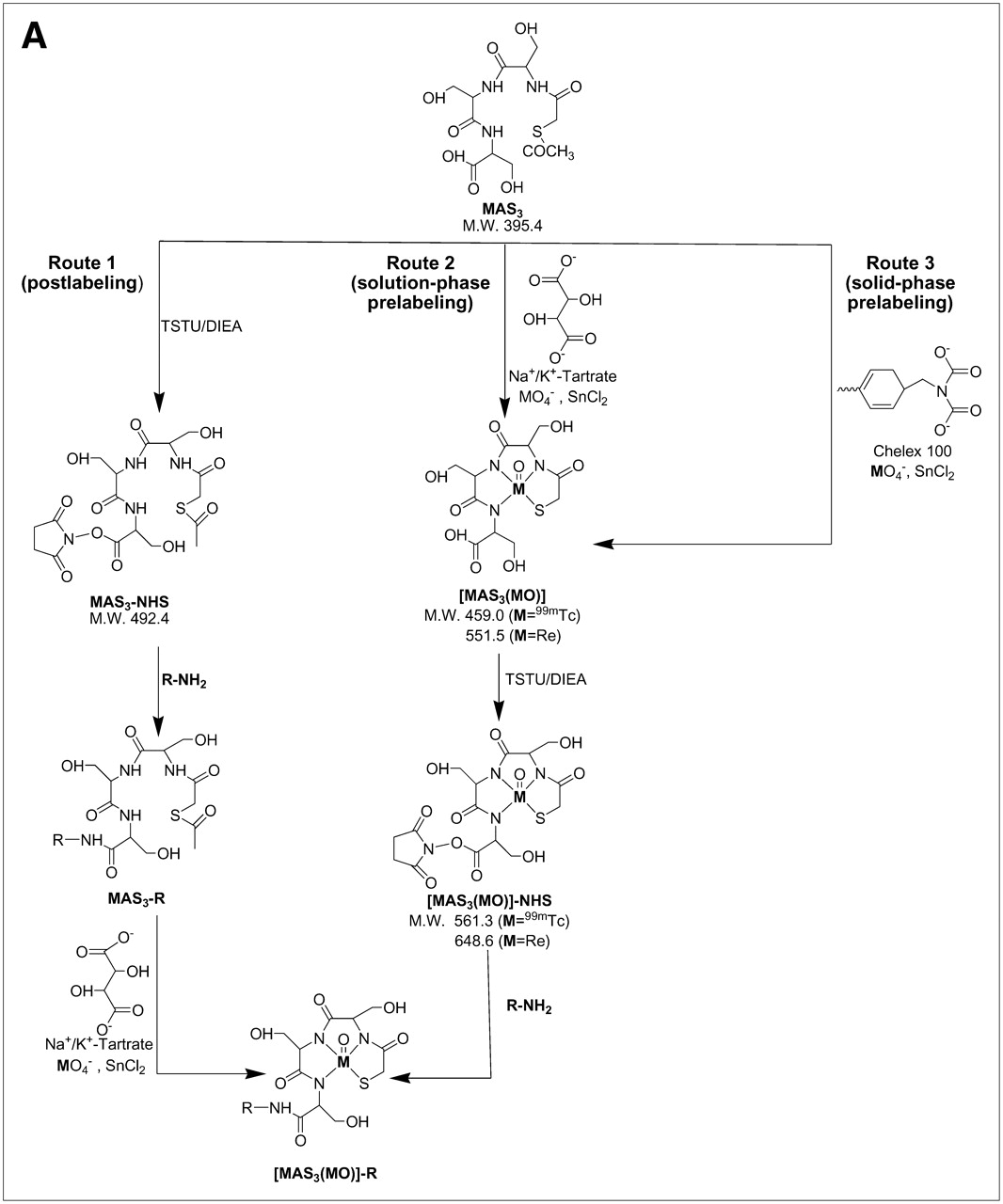
Unfortunately, highly anionic small molecules such as IV are difficult to use as 99mTc diagnostic radiotracers because the 12 net negative charges after conjugation to a chelator can potentially act as coligands for 99mTc. Furthermore, traditional pre- and postlabeling approaches for 99mTc (Fig. 2A) involve the use of excess quantities of an exchange ligand such as Na+/K+ tartrate. This creates 2 major problems. First, separation of 99mTc-tartrate away from a desired radiolabeled chelator is extremely difficult and requires high-resolution HPLC (Fig. 2B). For this reason, the radiolabeled tartrate peak is typically just accepted as “contaminant” and the entire labeling reaction is used (even in clinical settings). Second, nonradioactive exchange ligand is used in excess, hence, all subsequent chemical steps, such as solution-phase prelabeling (Fig. 2A) are competed by the exchange ligand's carboxylic acids, unless great care, and time-consuming separation, is used. Finally, if not purified to completion, nonradioactive tartrate can form reactive NHS esters and conjugate covalently to the targeting ligand, again requiring careful purification, and risking competition with radiotracer.

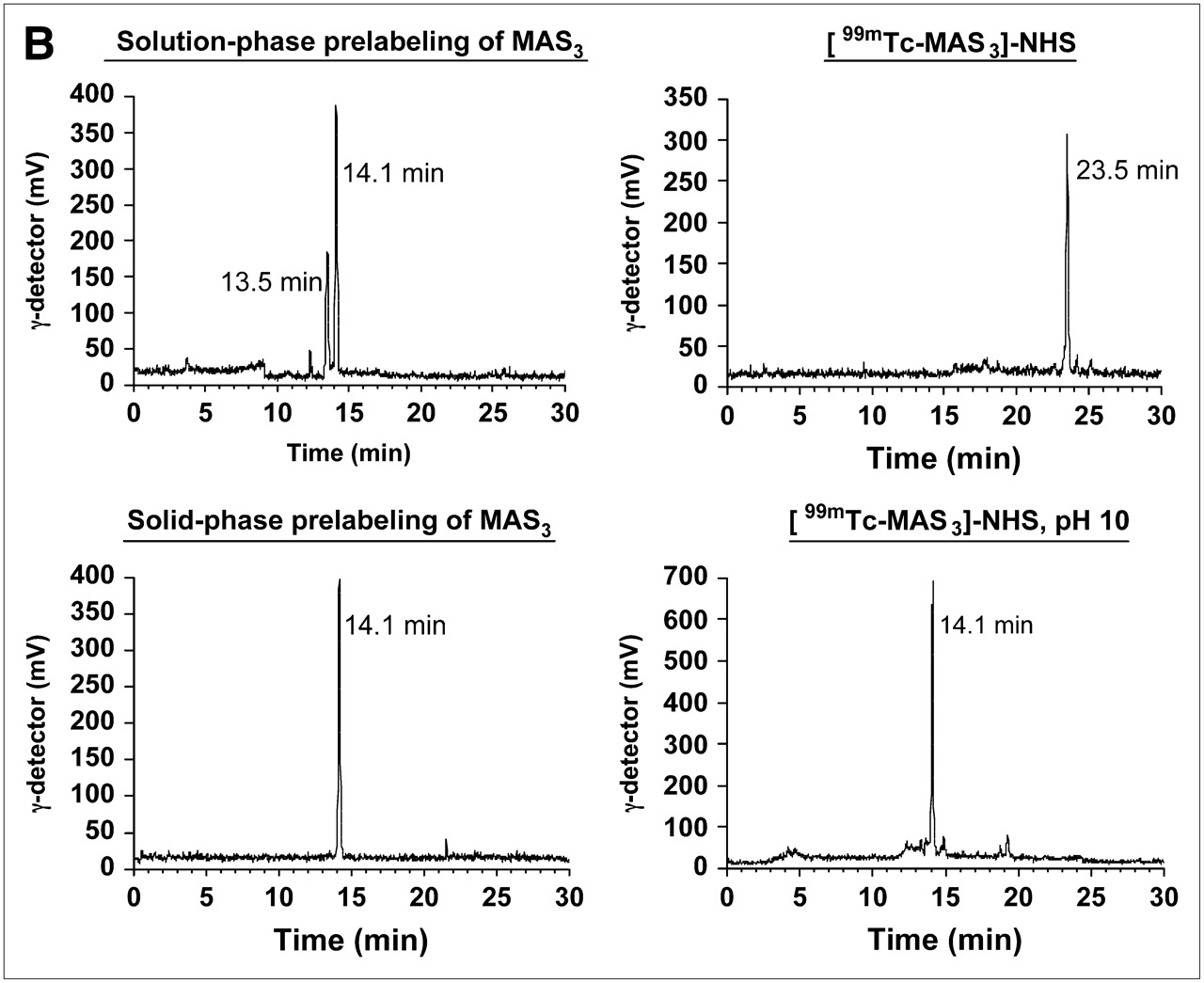
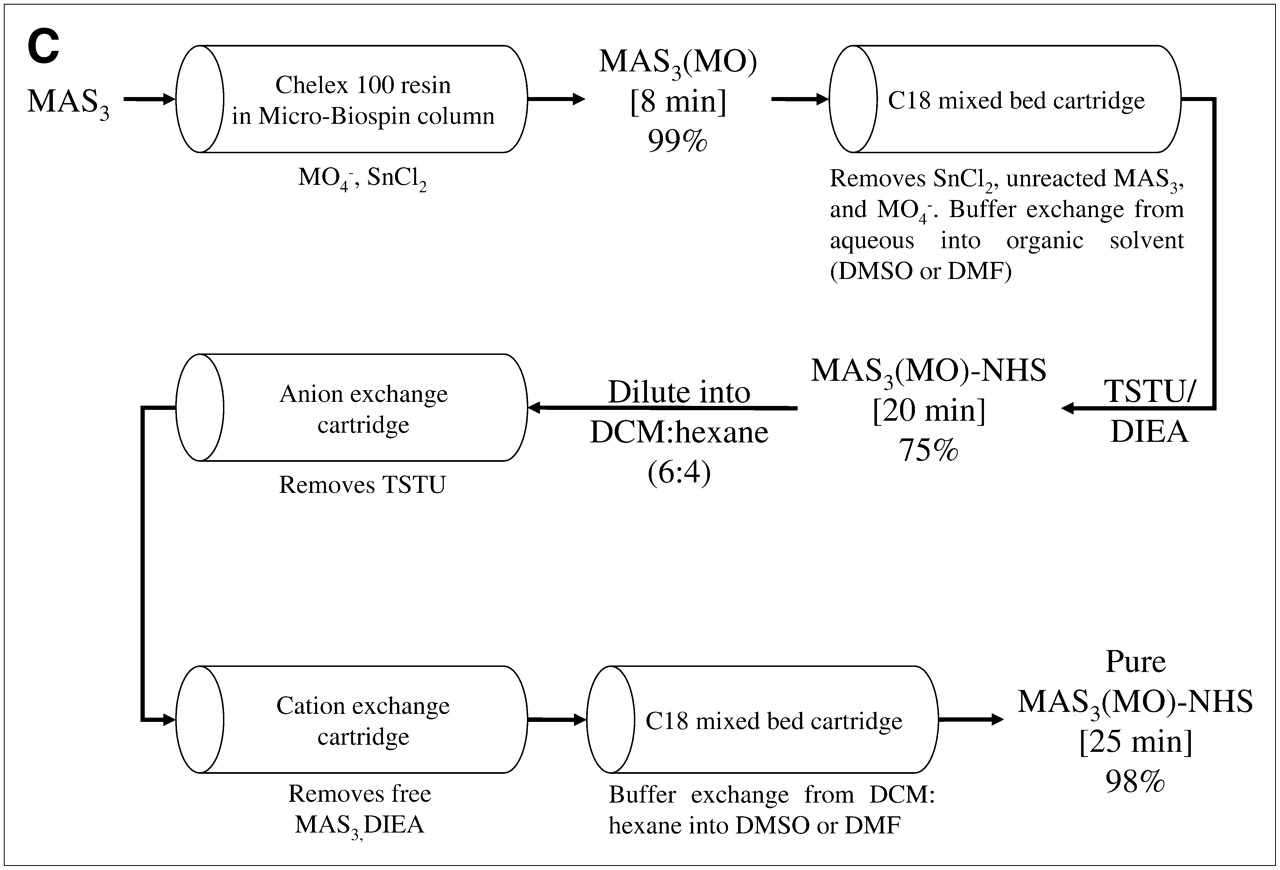
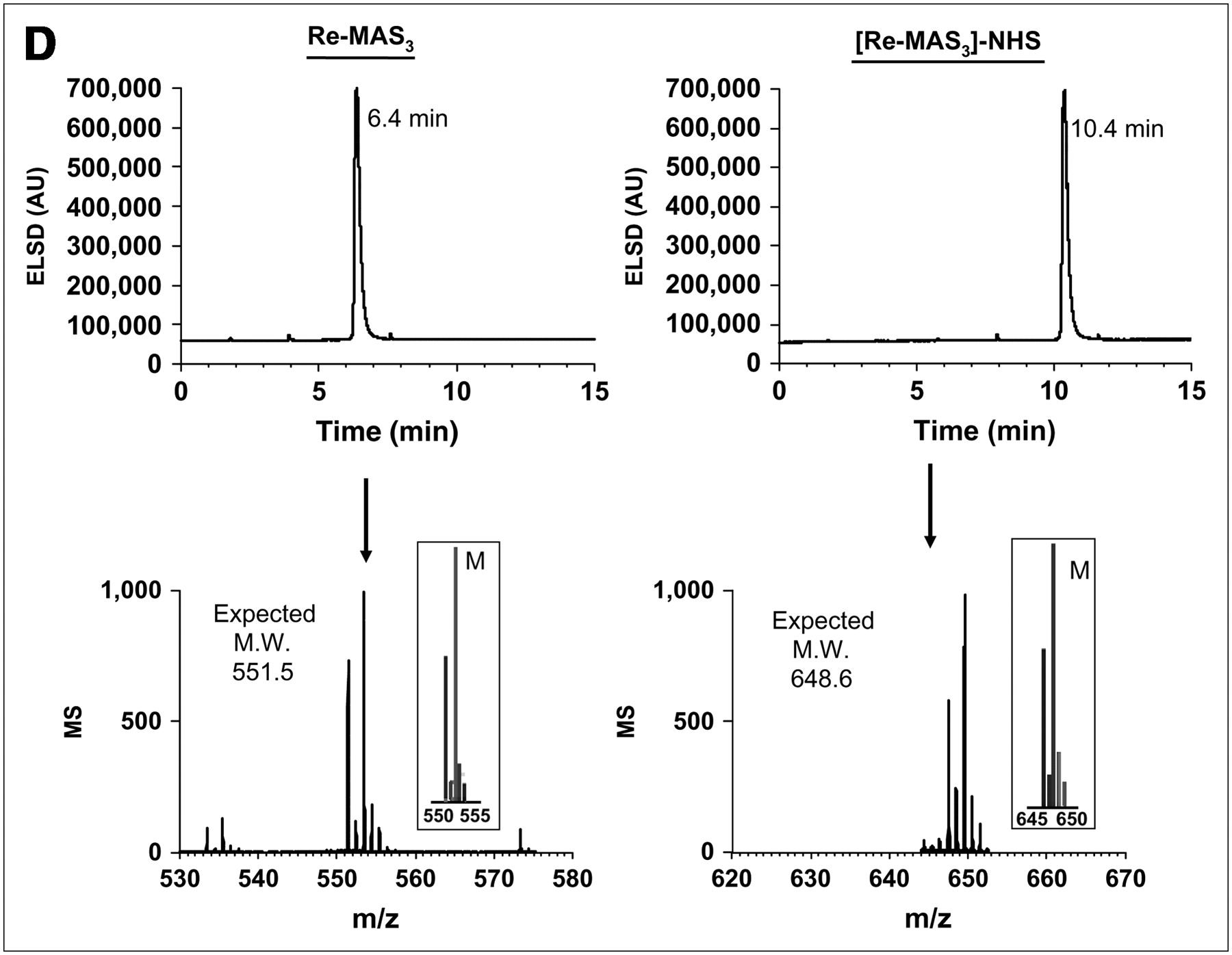
99mTc labeling strategies, solid-phase prelabeling, and purification of 99mTc-/Re-labeled MAS3 and MAS3-NHS. (A) General approaches for creating 99mTc-radiolabeled derivatives of small molecules and peptides include postlabeling (route 1) in presence of excess exchange ligand such as tartrate, solution-phase prelabeling using excess of exchange ligand (route 2), or solid-phase prelabeling (route 3) using Chelex 100 resin as described. (B) C18 HPLC radiochromatograms of conventional solution-phase prelabeling of 99mTc-MAS3 with excess tartrate (top left) and solid-phase prelabeling with Chelex 100 (bottom left). Solid-phase formation and purification of [99mTc-MAS3]-NHS (top right) and hydrolysis of NHS ester at pH 10 (bottom right). 99mTc-MAS3 and [99mTc-MAS3]-NHS elute at 14.1 and 23.5 min, respectively. 99mTc-Tartrate elutes at 13.5 min. (C) Simple, cartridge-based labeling and purification protocol to produce [99mTc-MAS3]-NHS is shown. Total elapsed time (brackets) and yield for each step are also shown. (D) C18 HPLC ELSD tracings (top) and mass spectra of major peak (bottom) for Re-MAS3 (left) and its NHS ester (right). Expected isotopic patterns for these Re derivatives are shown as insets. AU = absorbance units.
To solve the problems associated with traditional radiolabeling, we have developed a simple and rapid, cartridge-based method for converting readily available and inexpensive 99mTc-pertechnetate (Na99mTcO4) into a chemically pure, reactive NHS ester in organic solvent (Fig. 2C). The first key to this strategy is the elimination of soluble exchange ligand in favor of a solid-phase source of carboxylic acids (Chelex 100 resin). As described in detail earlier, only 8 min is required to fully load (99% radiochemical yield) a 99mTc chelator, such as MAS3 (9), with 99mTc (Fig. 2C). MAS3 was chosen over MAG3 and other 99mTc chelates given its low-background binding in primates after conjugation to targeting molecules (15).
The second key to this strategy is the transfer of 99mTc-MAS3 from aqueous buffer to nonaqueous buffer, as the final NHS ester is highly susceptible to attack by the hydroxyl ion of H2O. To accomplish this, and to remove unwanted molecules, the supernatant from the Chelex 100 resin is bound to a C18 mixed-bed cartridge, washed, and then eluted with desired organic solvent (DMSO or DMF). The NHS ester of 99mTc-MAS3 is then formed rapidly using TSTU and DIEA as described. Excess reactants are removed using tandem mixed-mode anion-exchange and cation-exchange cartridges, and the final product is concentrated on a C18 mixed-bed cartridge (Fig. 2C). Using this chemical strategy, [99mTc-MAS3]-NHS is prepared in ultrapure form (>99%; Fig. 2B) in DMSO or DMF, in only 25 min, with an overall radiochemical yield of 70%–75% relative to 99mTc-pertechnetate starting material. Reaction of the final product with basic water (pH 10) results in the rapid reformation of 99mTc-MAS3, thus proving the presence of an NHS ester (Fig. 2B). The typical specific activity of [99mTc-MAS3]-NHS produced using this strategy was 4.1 × 108 MBq/mmol (1.1 × 104 Ci/mmol).
To confirm, absolutely, the chemical structures produced, as well as to prepare nonradioactive derivatives of diagnostic agents for affinity measurements, the chemistry was repeated using Re. Although the exact procedure shown in Figure 2C worked well for Re-MAS3 chelates, the yield was low (≈20%), likely reflecting the difficulty others have had in preparing Re chelates (8). Hence, for preparative purification of Re-MAS3, solution-phase labeling was used, followed by formation and purification of [Re-MAS3]-NHS using our solid-phase approach. A high-resolution LC/MS HPLC system (7) was also used to verify that all cartridges performed as designed and to verify the purity of final compounds (Fig. 2D).
Solid-Phase Prelabeling of Highly Anionic PSMA Small Molecules
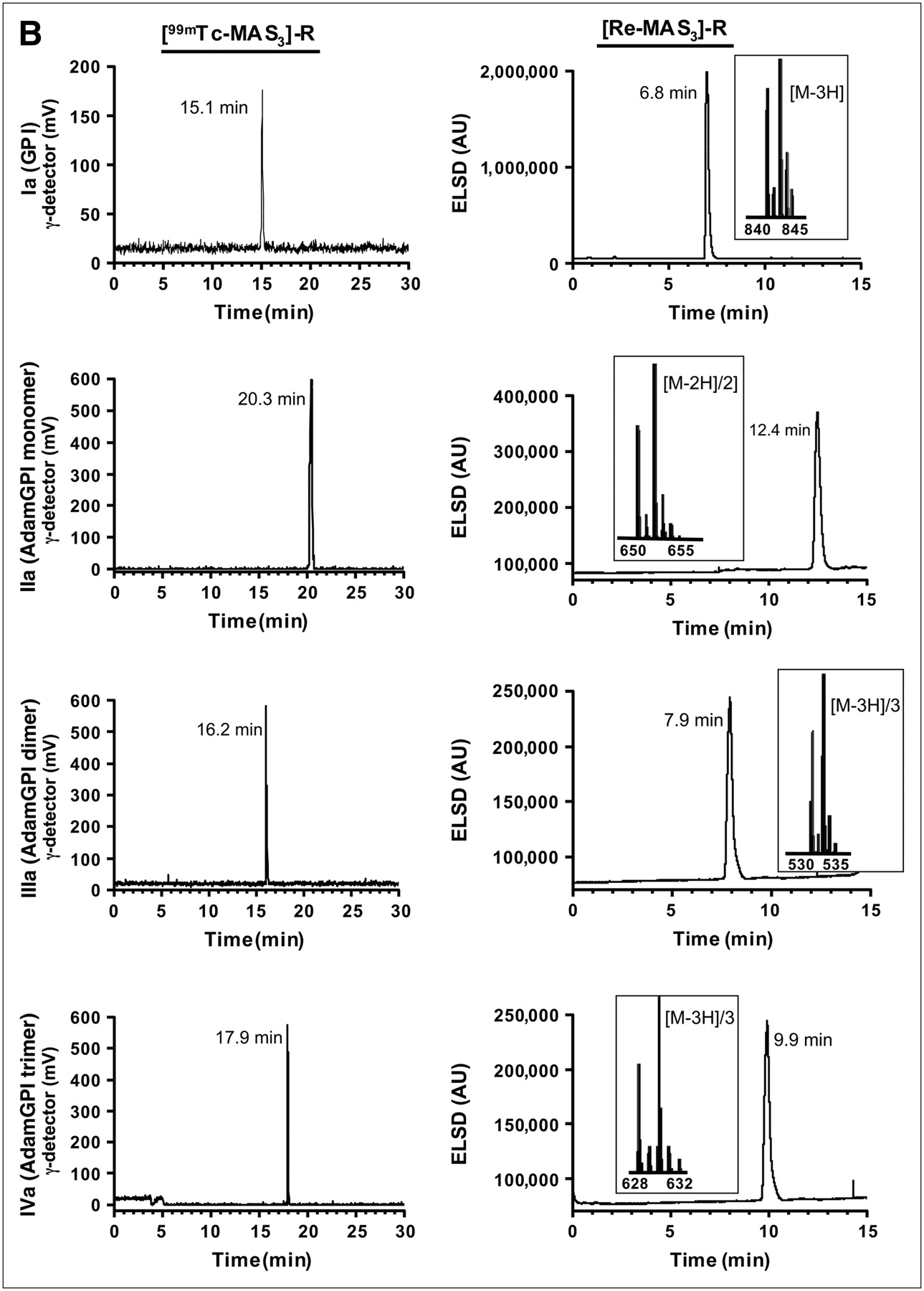
Using this solid-phase preloading strategy, GPI-containing molecules of increasing ligand valency were labeled with either 99mTc or Re in 1 chemical step as shown in Figure 3A. For 99mTc-labeled molecules, no further purification was necessary, as the NHS ester reaction with an amine in dry DMSO or DMF goes to completion without side reactions (Fig. 3B). For Re-labeled molecules, which were to be used for affinity measurements, LC/MS analysis with ELSD (7) was used to ensure the highest possible purity and to confirm the expected isotopic pattern (Fig. 3B).
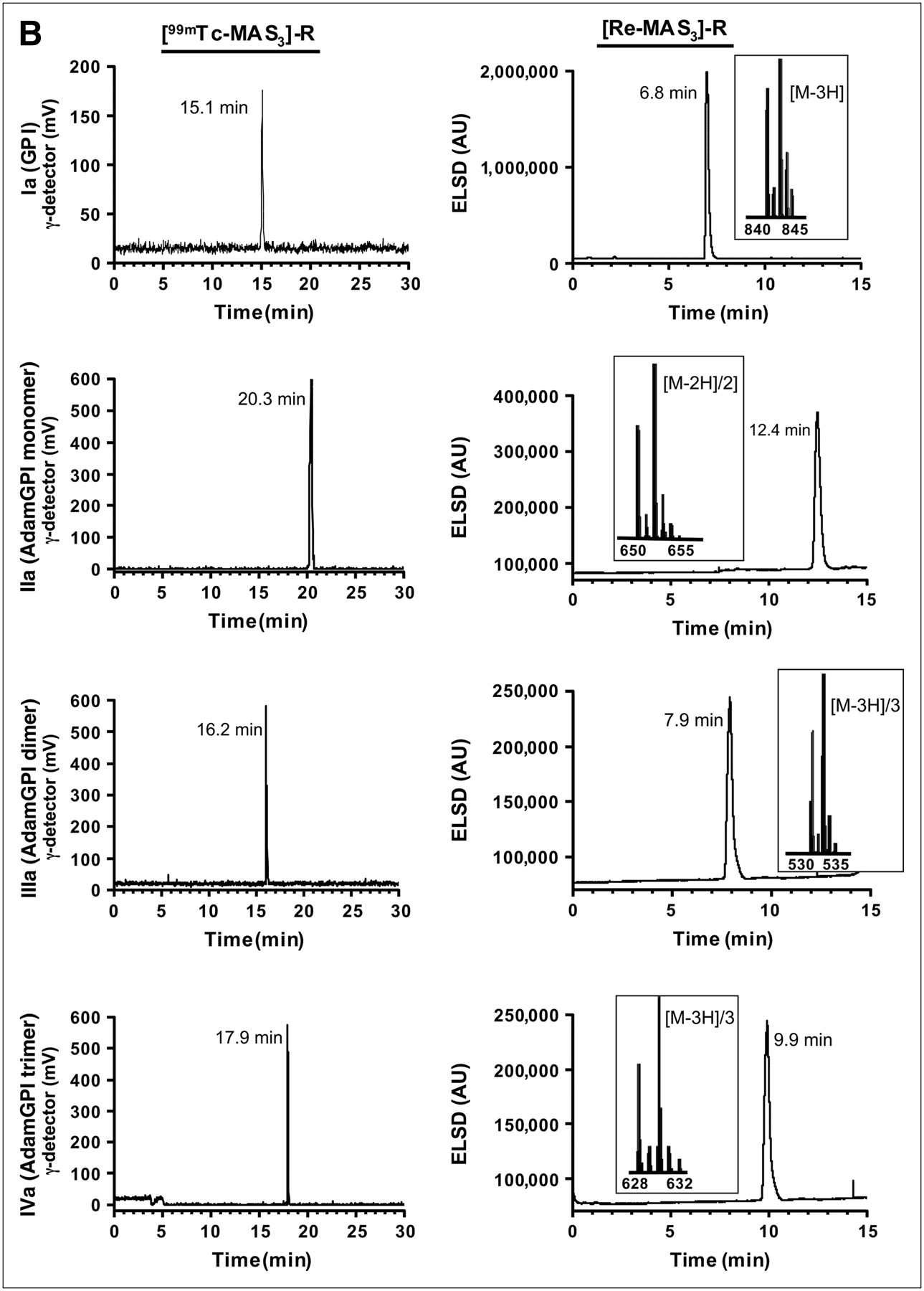

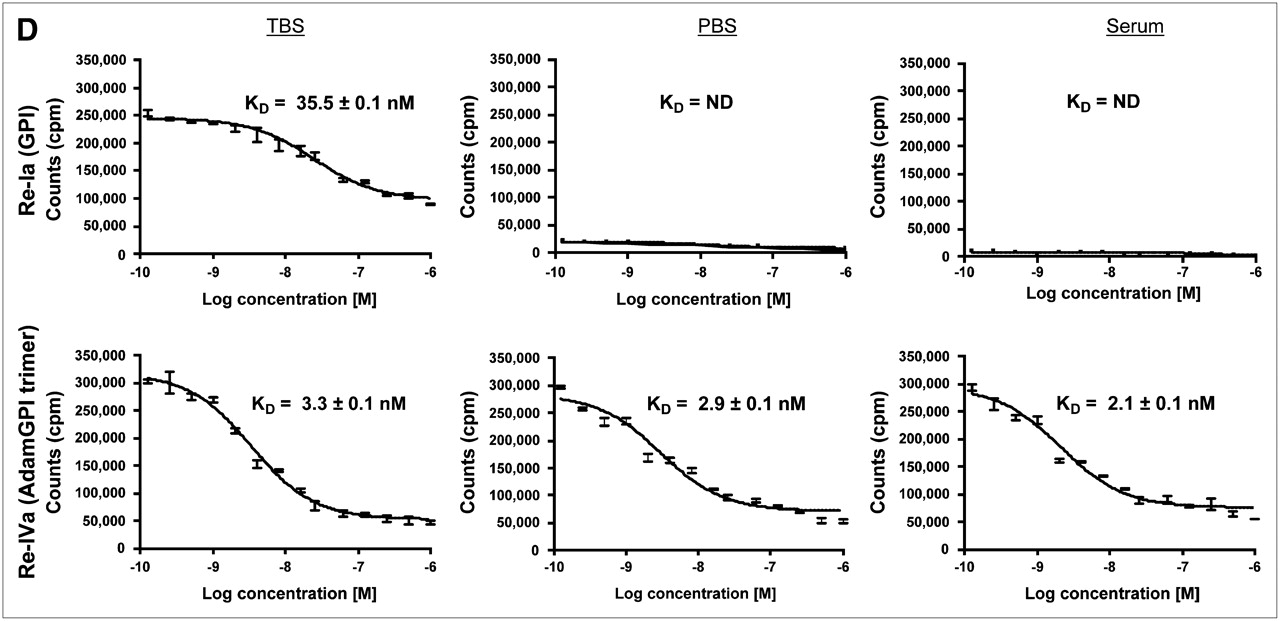
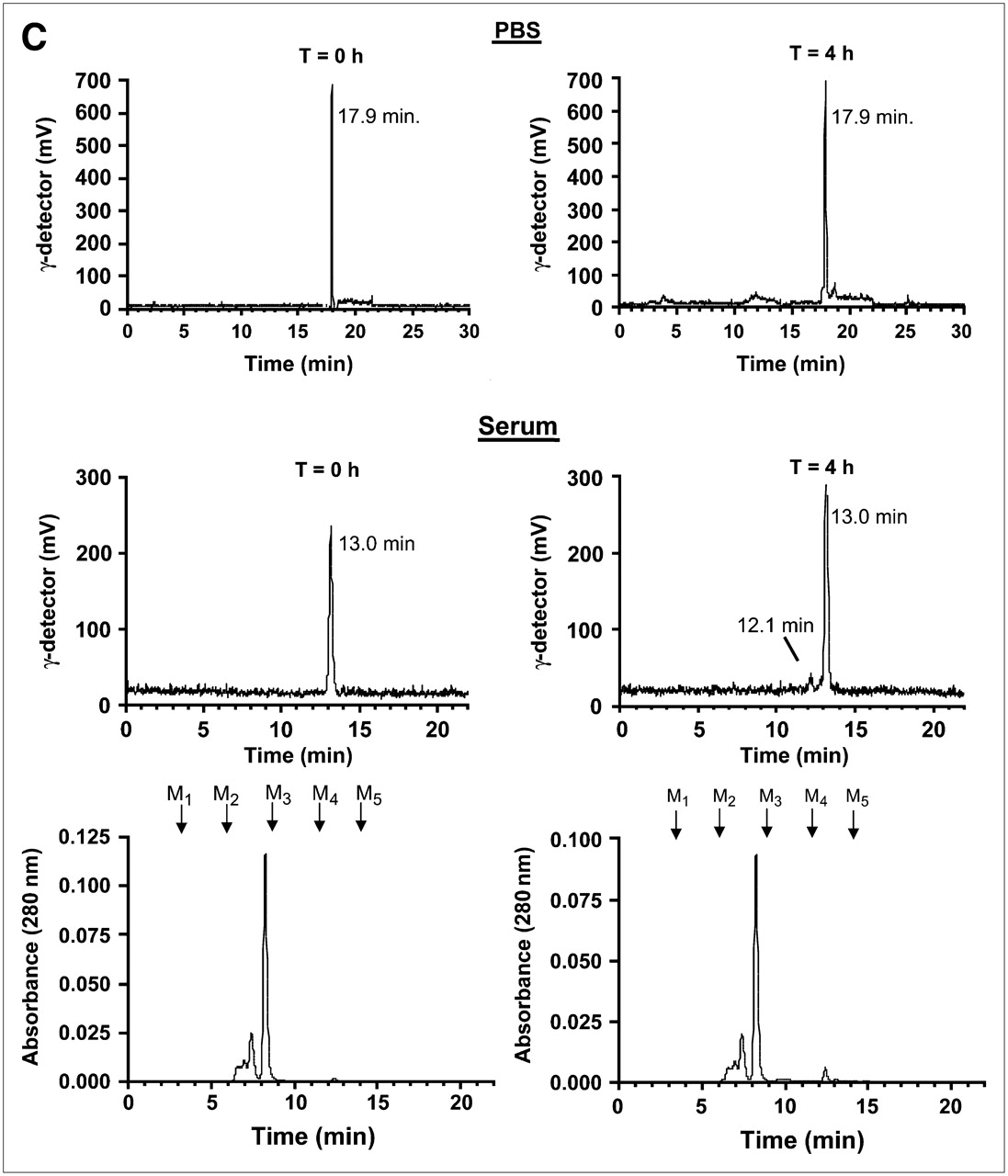
Preparation, purification, and analysis of PSMA-specific radiotracers. (A) Single nucleophiles (primary amines) of small molecules from Fig. 1 were conjugated in 1 step to [99mTc-MAS3]-NHS using solid-phase preloading strategy described in text to create PSMA-specific radiotracers. (B) RP-HPLC analysis of compounds I–IV after solid-phase prelabeling using either 99mTc (left) or Re (right). Rt values for compounds Ia–IVa are shown, as are ES-TOF(−) mass spectra of peak for Re compounds (insets). AU = absorbance units. For comparison, Rt values for [99mTc-MAS3]-NHS and [Re-MAS3]-NHS on their optimized gradients are 23.5 and 10.4 min, respectively (Figs. 2B and 2D). (C) Serum protein binding and serum stability of solid-phase prelabeled 99mTc PSMA-specific small molecules. Compound 99mTc-IVa was incubated for 0 or 4 h at 37°C in PBS (top) or 100% serum (bottom) and then subjected to HPLC analysis. Samples in PBS were resolved on a Symmetry C18 column. Samples in serum were resolved on a 120-Å high-resolution gel-filtration column and also include absorbance (280 nm) tracings. Rt values for gel-filtration markers M1−M5 are shown as arrows. Marker Rt values were M1 = 6.6 min, M2 = 8.2 min, M3 = 9.2 min, M4 = 11.1 min, and M5 = 13.4 min. (D) Live cell-binding assay. PSMA-positive LNCaP cells grown on 96-well filter plates as described in text were incubated for 20 min at 4°C with monomeric 99mTc-Ia (top row) or trimeric 99mTc-IVa (bottom row) in presence of increasing concentrations of homologous nonradioactive test compound. Shown are results for monomeric Re-Ia (top row) and trimeric Re-IVa (bottom row) in TBS (left), PBS (middle), and 100% serum (right). Also shown are mean affinities ± 95% confidence intervals for 3 independent assays. N.D. = none detected.
The stability of adamantine-trimerized GPI molecule (IVa) was tested by incubation at 37°C for 4 h in either PBS or 100% serum. HPLC analysis (Fig. 3C) revealed 100% stability in PBS and 99% stability in serum, with 1% appearing as an early-eluting peak.
Bioactivity and Radioscintigraphic Imaging of PSMA-Specific Radiotracers
Using the nonradioactive Re derivatives as test compounds, 99mTc derivatives as radiotracers, and a high-throughput 96-well filter plate assay (7), the absolute affinity of each molecule for the surface of living prostate cancer cells was measured using homologous competition (Table 1). Actual raw data from the assay using monomeric [99mTc-MAS3]-GPI (Ia) as radiotracer (Fig. 3D, top) and trimeric [99mTc-MAS3]-AdamGPI (IVa) as radiotracer (Fig. 3D, bottom) confirm that both phosphate and serum compete effectively with monomeric radiotracers for the active site of PSMA but are unable to compete with trimeric radiotracers, resulting in nearly identical affinities of IVa under all physiologic conditions. The measured maximum number of binding sites (Bmax) values for compounds Ia/IIa, IIIa, and IVa were 1.6 × 105, 2.1 × 105, and 2.6 × 105 PSMA receptors per cell, respectively, which is consistent with previously published reports of 1.8 × 105 to 8.0 × 105 for PSMA-specific monoclonal antibodies (16,17).
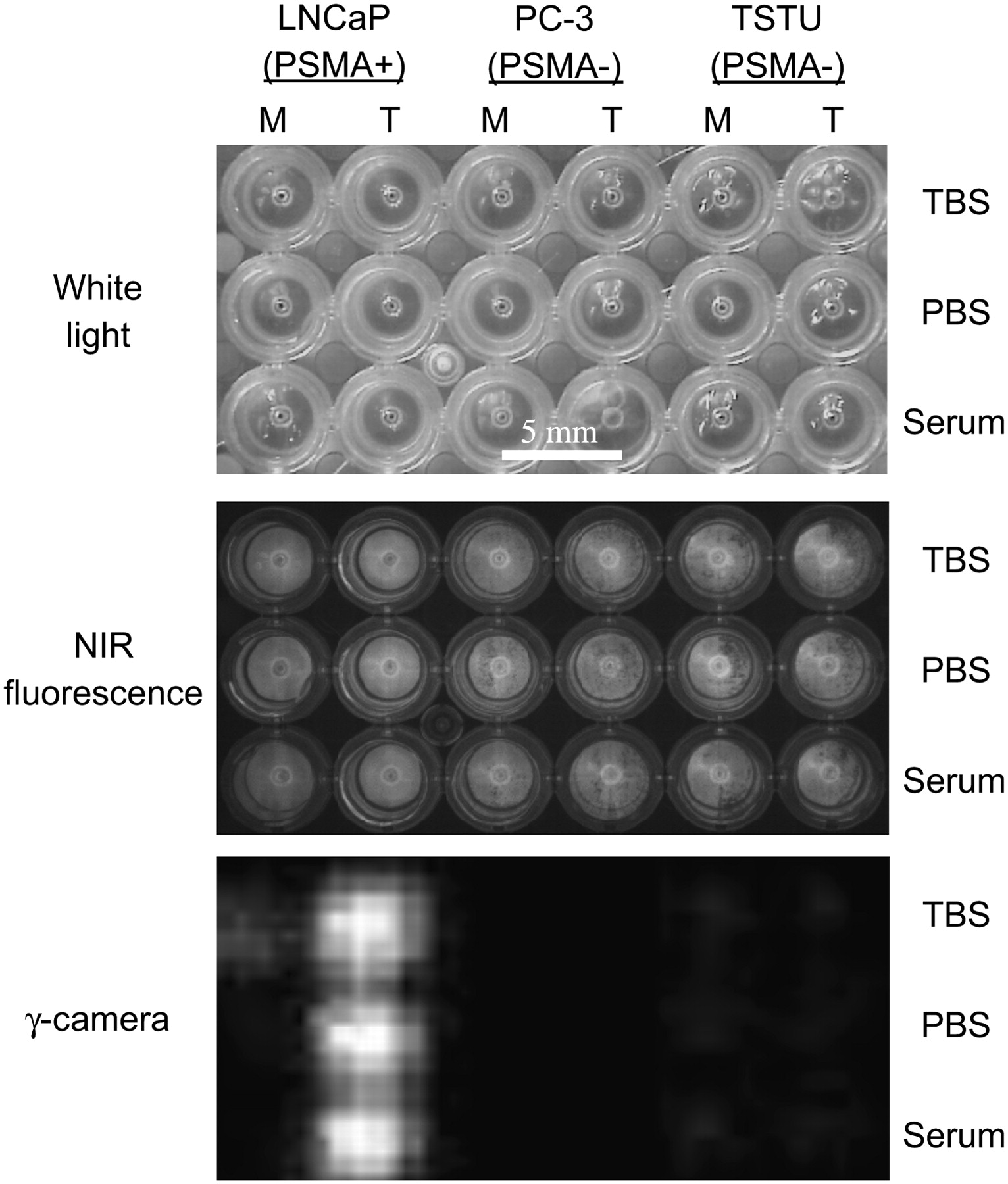
To confirm that the synthesized radiotracers permit sensitive and specific radioscintigraphic imaging of prostate cancer cells, approximately 35,000 PSMA-positive (LNCaP) or PSMA-negative (PC-3 and TsuPR1) cancer cells were grown in each well of a 96-well filter plate and incubated at 4°C for 20 min with the indicated tracer. As shown in Figure 4, and confirming the results in Table 1 and Figure 3D, 99mTc-labeled trimeric AdamGPI (IVa) bound with high affinity to living prostate cancer cells expressing PSMA in all buffers, including 100% serum, and no binding was detectable on PSMA-negative cells.

Radioscintigraphic imaging of 99mTc-labeled PSMA-specific radiotracers: Live cell binding of 99mTc-Ia (GPI monomer; M) and 99mTc-IVa (AdamGPI trimer, T) was performed in TBS (top row), PBS (middle row), and 100% serum (bottom row) for 20 min at 4°C using PSMA-positive LNCaP cells (left), PSMA-negative PC-3 cells (middle), and PSMA-negative TsuPR1 cells (right) grown on 96-well filter plates, followed by extensive washing. Cells were independently loaded with NIR fluorophore IR-786 to assess viability and confluence. Shown are white light (top), NIR fluorescence (middle), and γ-detector (bottom) images of cells grown on 96-well filter plates.
DISCUSSION
Patients with prostate and other cancers are in desperate need of targeted diagnostic and therapeutic agents. One common strategy for the development of such agents is to use a small molecule or peptide-targeting ligand, conjugated to a desired functional molecule (e.g., contrast agent, radiotracer, or therapeutic) via an isolating linker (7). This robust strategy requires conjugation chemistry that is simple and rapid. Although other pre- and postlabeling strategies for preparing 99mTc SPECT radiotracers have been described (8,18), none has the combined features of taking <1 h with inexpensive disposable cartridges, eliminating exchange ligand contaminants, producing ultrapure reactive NHS esters in organic solvent, producing high-specific-activity radiotracers, producing a stable amide bond, and eliminating the need for HPLC purification of final labeled agents. Eliminating the need for any HPLC purification steps makes the strategy amenable to high-throughput small molecule and peptide screening and will also hopefully enable SPECT—especially, preclinical SPECT—by investigators who would otherwise not have access to the expensive equipment and chemistry expertise needed with other approaches. Another key feature is that it begins with 99mTc-pertechnetate, an inexpensive reagent available at virtually every medical center. Finally, our solid-phase strategy is inherently automatable.
An important general limitation of NHS ester chemistry is that the hydroxyl ion of water is a potent competitor. NHS esters of fluorophores are typically used in millimolar concentrations and in excess of the targeting molecule. This is not true of radioactive NHS esters, such as [99mTc-MAS3]-NHS, which are used in nanomolar concentrations. Hence, the use of [99mTc-MAS3]-NHS in aqueous buffer will result in low labeling efficiencies. When used in dry organic solvent, however, [99mTc-MAS3]-NHS reacts rapidly and completely with primary amines. Another potential limitation is the availability of the nucleophile within the targeting ligand. Each of our GPI derivatives has been engineered to have a flexible 6-carbon spacer; however, more sterically hindered amines could be difficult to conjugate.
Even with careful chemical engineering, small molecule and peptide-targeting ligands often have poor affinity or are competed by endogenous ligands. A general approach to solving these problems was proposed by Whitesides' group (13) and involves multimerizing binding epitopes into an optimally spaced rigid structure. To accomplish this, we have created a tri-NHS adamantane derivative (14), which permits the conjugation of up to 3 (same or different) targeting ligands. The molecule also has an isolating linker and a deprotectable primary amine for subsequent conjugation to contrast agents, radiotracers, or therapeutics. Adamantane, being the core structure in diamond, has maximal rigidity. In this study, we conjugated GPI through a propionyl acid spacer and made no attempt to optimize intermolecular spacing or overall rigidity. Future studies will focus on these issues.
Nonetheless, the results to date are encouraging. First, conjugation of a single GPI monomer to a single adamantane core had only a negligible effect on affinity (Table 1; compare Ia with IIa). Second, increasing multivalency from 1 to 3 led to dramatic improvements in both affinity and competition with endogenous anions. Indeed, affinity not only increased almost 2 logarithms when measured in TBS, but as a trimer was unaffected by serum (Table 1). The results also reveal, however, that the GPI molecules are not as isolated from the effect of metal chelate as one would like. For example, when Re-MAS3 is conjugated to either GPI monomer or AdamGPI Trimer, there is a 3- to 8-fold decrease in affinity. Nevertheless, the final affinity of IVa (3 nM) is equivalent to that of previously described PSMA monoclonal antibodies (16).
Because it is known that PSMA undergoes both constitutive and inducible endocytosis (19), we were careful to perform all affinity measurements on living cells at 4°C. However, both NIR fluorescent GPI derivatives (2) and 99mTc-labeled derivatives (data not shown) are concentrated inside cells via endocytosis, which serves to “amplify” radiotracer signal over time. This important feature of PSMA will likely improve in vivo performance and also needs to be incorporated into pharmacokinetic modeling.
CONCLUSION
Trimeric GPI conjugated to 99mTc-MAS3 (IVa) may someday be a valuable diagnostic agent for clinical use. It has a 3 nM affinity for the surface of living prostate cancer cells in serum; 99% of it is unchanged after incubation in warm serum for 4 h; and covalent conjugation to 99mTc-MAS3 is performed in 1 step without the need for subsequent purification. Ongoing experiments will characterize its biodistribution and clearance in vivo.
Acknowledgments
We thank Pavel Majer, Takashi Tsukamoto, and Barbara Slusher from MGI Pharma for supply of the GPI compound, Jürgen T. Renze for initial preparation of MAS3, J. Anthony Parker for many helpful discussions, Barbara L. Clough for editing, and Grisel Vazquez for administrative assistance. This work was supported by NIH grant R01-CA-115296, grant DFG MA 2529/3 from the Deutsche Forschungsgemeinschaft, and grants from the Lewis Family Fund and the Ellison Foundation.
Footnotes
-
COPYRIGHT © 2007 by the Society of Nuclear Medicine, Inc.
References
- Received for publication January 31, 2007.
- Accepted for publication April 23, 2007.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- A Bibliometric Analysis of Radiologic Technology (1963-2011)
- Effect of Small-Molecule-Binding Affinity on Tumor Uptake In Vivo: A Systematic Study Using a Pretargeted Bispecific Antibody
- Assessing Antibody Pharmacokinetics in Mice with In Vivo Imaging
- Targeting Prostate Cancer Cells In Vivo Using a Rapidly Internalizing Novel Human Single-Chain Antibody Fragment
- Annexin A2 Is a Molecular Target for TM601, a Peptide with Tumor-targeting and Anti-angiogenic Effects
- New Technologies for Human Cancer Imaging
- Quantitation of CXCR4 Expression in Myocardial Infarction Using 99mTc-Labeled SDF-1{alpha}