


Abstract
Vascular endothelial growth factor A (VEGF-A) and its receptors, Flt-1/FLT-1 (VEGFR-1) and Flk-1/KDR (VEGFR-2), are key regulators of tumor angiogenesis and tumor growth. The purpose of this study was to determine the antiangiogenic and antitumor efficacies of a vasculature-targeting fusion toxin (VEGF121/rGel) composed of the VEGF-A isoform VEGF121 linked with a G4S tether to recombinant plant toxin gelonin (rGel) in an orthotopic glioblastoma mouse model by use of noninvasive in vivo bioluminescence imaging (BLI), MRI, and PET. Methods: Tumor-bearing mice were randomized into 2 groups and balanced according to BLI and MRI signals. PET with 64Cu-1,4,7,10-tetraazacyclododedane-N,N′,N″,N‴-tetraacetic acid (DOTA)-VEGF121/rGel was performed before VEGF121/rGel treatment. 18F-Fluorothymidine (18F-FLT) scans were obtained before and after treatment to evaluate VEGF121/rGel therapeutic efficacy. In vivo results were confirmed with ex vivo histologic and immunohistochemical analyses. Results: Logarithmic transformation of peak BLI tumor signal intensity revealed a strong correlation with MRI tumor volume (r = 0.89, n = 14). PET with 64Cu-DOTA-VEGF121/rGel before treatment revealed a tumor accumulation (mean ± SD) of 11.8 ± 2.3 percentage injected dose per gram at 18 h after injection, and the receptor specificity of the tumor accumulation was confirmed by successful blocking of the uptake in the presence of an excess amount of VEGF121. PET with 18F-FLT revealed significant a decrease in tumor proliferation in VEGF121/rGel-treated mice compared with control mice. Histologic analysis revealed specific tumor neovasculature damage after treatment with 4 doses of VEGF121/rGel; this damage was accompanied by a significant decrease in peak BLI tumor signal intensity. Conclusion: The results of this study suggest that future clinical multimodality imaging and therapy with VEGF121/rGel may provide an effective means to prospectively identify patients who will benefit from VEGF121/rGel therapy and then stratify, personalize, and monitor treatment to obtain optimal survival outcomes.
Glioblastoma multiforme (GBM) is a grade IV primary brain tumor with a poor prognosis despite advances in surgical intervention, chemotherapy, and radiotherapy over the last 30 years (1). Histologic hallmarks of GBM include necrotic regions surrounded by anaplastic cells, vascular proliferation reflecting tumor ischemia, and active angiogenesis (2). Effective therapies for GBM, and brain tumors in general, have been difficult to develop because of the lack of targetable tumor-specific markers. The poor outcomes for patients with GBM and other malignant brain tumors warrant improved therapies and complementary diagnostic imaging to monitor and optimize treatment strategies.
Angiogenesis in tumor growth, invasion, and metastasis has been a major focus of recent cancer research, prompting the development of numerous antiangiogenic therapies. Proteins regulating angiogenesis include vascular endothelial growth factor (VEGF) A (VEGF-A), tumor necrosis factor-α, epidermal growth factor, hypoxia-inducible factors, and interleukin 8 (3). The receptors for these proteins have been demonstrated in both animal models (4) and clinical studies (5) to be upregulated on tumor vasculature, thus providing therapeutic targets that bypass the tumor parenchyma. In particular, VEGF-A has been well studied for its prominent role in regulating tumor angiogenesis (6), along with its receptors, FLT-1 (VEGFR-1) and KDR (VEGFR-2) in humans and Flt-1 and Flk-1 in mice (7). These receptors are expressed in various solid neoplasms, including, but not limited to, bladder (8), breast (9), and pancreatic (10) tumors. GBM is a prime example of VEGF-induced tumor neovascularization, as the upregulation of VEGFR-2 on tumor vasculature and the overexpression of VEGF-A ligand by tumor cells have been implicated as poor prognostic markers for gliomas (11).
FLT-1 and KDR are prime targets for antiangiogenic therapies because they are upregulated on tumor vascular endothelium but not on surrounding normal quiescent vessels (12). Studies have shown that VEGFR-2 is primarily responsible for the proangiogenic effects of VEGF in solid tumors (13). Tyrosine kinase inhibitors, monoclonal antibodies, ribozymes, small interfering RNA, and soluble receptors that block the VEGF signaling cascade are all currently being developed and tested (13,14). We previously characterized a novel 84-kDa vasculature-targeting fusion protein (VEGF121/rGel) composed of the VEGF-A isoform VEGF121 linked with a G4S tether to recombinant plant toxin gelonin (rGel) (15). The VEGF121/rGel fusion toxin has been shown to be highly specific and cytotoxic for both quiescent and dividing porcine aortic endothelial (PAE) cells expressing VEGFR-2 (PAE/KDR) but not for PAE cells expressing VEGFR-1 (PAE/FLT-1) (15). VEGF121/rGel has also been shown to completely suppress ocular neovascularization (16) and inhibit the growth of melanomas as well as prostate (15), breast (17), and bladder (8) tumors in mouse models with low systemic toxicity.
There are large variations in the expression of the VEGF-A ligand in solid tumors and receptor modulation in developing tumor neovasculature, and little is known about the precise in vivo fluctuations of VEGF-related content (18). Therefore, a critical step in future clinical applications of VEGF121/rGel and other antiangiogenic therapies is the development of effective noninvasive in vivo imaging techniques for monitoring VEGF and VEGFR expression as well as treatment efficacy. The purpose of this study was to evaluate the treatment efficacy of vasculature-targeting fusion toxin VEGF121/rGel in an orthotopic glioblastoma mouse model by use of noninvasive in vivo multimodality molecular imaging. Our results demonstrated that VEGF121/rGel specifically targets and damages tumor neovasculature, resulting in decreased tumor DNA synthesis, increased apoptosis, and overall decreased glioblastoma growth. Tumor growth changes can be monitored by use of bioluminescence imaging (BLI) and MRI. Treatment-induced alterations in the DNA proliferative index can be analyzed with 18F-flurothymidine (18F-FLT) (19). Future clinical diagnostic imaging combined with therapy with VEGF121/rGel may provide an effective means of monitoring and personalizing the treatment of glioblastoma and other tumors with a neovascular tree expressing VEGFR-2 and may clarify mechanisms of resistance to antiangiogenic therapies.
MATERIALS AND METHODS
64Cu Labeling of DOTA-VEGF121/rGel
The synthesis, expression, and purification of the fusion toxin VEGF121/rGel were conducted as previously described (15). 1,4,7,10-Tetraazacyclododedane-N,N′,N″,N‴-tetraacetic acid (DOTA) conjugation was performed as previously described with slight modifications and a reaction ratio of DOTA to VEGF121/rGel of 100:1 (20,21). DOTA-VEGF121/rGel was purified by use of a PD-10 desalting column (GE Healthcare) and concentrated by use of a Centricon apparatus (Millipore). 64CuCl2 (74 MBq) was diluted in 300 μL of 0.1N sodium acetate buffer (pH 6.5) and then added to 50 μg of DOTA-VEGF121/rGel. The reaction mixture was incubated for 1 h at 40°C with constant shaking. 64Cu-DOTA-VEGF121/rGel was then purified by use of a PD-10 column with phosphate-buffered saline (PBS) as the mobile phase. The average number of DOTA chelator molecules per VEGF121/rGel molecule was determined as previously described (20).
Cell-Binding Assay and Functional Assay
A cell-binding assay with VEGF121/rGel and the DOTA-VEGF121/rGel conjugate was performed as previously described with 125I-VEGF165 (specific activity, 74 TBq/mmol; GE Healthcare) as the radioligand (22). The best-fit 50% inhibitory concentrations (IC50s) for the PAE/KDR cells were calculated by fitting the data with GraphPad Prism (GraphPad Software, Inc.). Experiments were performed twice with triplicate samples.
Details of the functional assay were reported earlier (22). The cell lysate was immunoblotted with an antiphosphotyrosine antibody (Santa Cruz Biotechnology) for phosphorylated KDR to compare the functional activities of DOTA-VEGF121/rGel and VEGF121/rGel. Tubulin was used as the loading control.
Orthotopic Glioblastoma Xenografts and Treatment Protocol
All animal procedures were performed according to a protocol approved by the Stanford University Administrative Panels on Laboratory Animal Care. Athymic nude mice (nu/nu; Harlan) at 4–6 wk of age were given intracranial injections in the right frontal lobe at coordinates 2 mm lateral and 0.5 mm anterior from the bregma and 2.5 mm intraparenchymally (23). Each mouse was injected with 105 firefly luciferase transfected U87MG human glioblastoma cells (U87MG-fLuc) suspended in 5 μL of PBS. Tumor cells were allowed to engraft for 7 d, at which point in vivo BLI and MRI were performed weekly to assess tumor growth. On day 34 after inoculation, BLI and MRI were conducted to assess tumor signal intensity and tumor volume before treatment. Mice were randomized to 2 groups: control (n = 6) and VEGF121/rGel treatment (n = 9). A maximum tolerated dose (MTD) of 45 mg/kg for VEGF121/rGel was previously established (17), and ∼50% of the MTD was used for treatment purposes (120 μg × 4 = 480 μg per mouse; 24 mg/kg administered every other day for a total of 4 doses). Saline administration was used as a single control on the basis of previous studies that demonstrated no impact on tumor growth when the same doses of rGel alone were administered (8,15). It was also believed that no therapeutic advantages would be gained by using free VEGF121 as a control, as it would most likely induce, rather than inhibit, angiogenesis. Preliminary work in our laboratory has also shown no effects of rGel alone on subcutaneous U87MG glioblastoma growth (data not shown).
Treatment with VEGF121/rGel began on day 35 after inoculation, the time at which previous experiments showed that tumor angiogenesis and growth began to increase exponentially (23). For treatment, the animals in the treatment group each received a 120-μL intraperitoneal injection of VEGF121/rGel (1 mg/mL) every other day for a total of 4 treatments; the control animals each received equivalent injections of saline. The weights of all of the animals were recorded every other day to monitor potential toxicity effects. The animals were sacrificed after 4 doses of VEGF121/rGel and perfused with 20 mL of cold saline and then 20 mL of 10% formalin. The brains were then embedded in paraffin for histologic analysis.
BLI
Intracranial tumor growth bioluminescence was assessed by use of a Xenogen IVIS 200 small-animal imaging system (Xenogen Corp.) (23). For BLI, an average of ten 1-min-exposure bioluminescence acquisitions were collected between 0 and 40 min after substrate injection to confirm the peak photon emission recorded as the maximum photon efflux per second. Identical illumination settings (lamp voltage, filters, f-stop, field of view, binning, excitation filter block, and emission filter open) were used to acquire all images throughout the study. Data were analyzed by use of total photon flux emission (photons per second) in a region of interest covering the entire brain. BLI was conducted on a weekly basis after tumor inoculation and at 24 h after the administration of each VEGF121/rGel dose to evaluate treatment progression.
MRI
Intracranial tumor growth was confirmed by gadolinium-enhanced MRI with a 4.7-T small-animal MRI system (Omega, GE Healthcare) equipped with a volume-based transmit/receive coil and an inner diameter of 4 cm (23). Tumor volume was assessed by use of the free-hand region-of-interest function of Image J software (National Institutes of Health). MRI reconstruction of tumor volume has been shown to have a strong correlation (r = 0.96) with traditional histologic reconstruction (24), with MRI having the disadvantage of partial-volume effects and the histologic technique having problems with tissue loss and shrinkage.
PET Imaging
The details of our PET image acquisition and quantification procedure were previously described (25–27). Images were reconstructed by use of a 2-dimensional ordered-subsets expectation maximum algorithm with no attenuation or scatter correction (28). Mice were intravenously injected with 5–10 MBq of 64Cu-DOTA-VEGF121/rGel or 18F-FLT via the tail vein under 1%–2% isoflurane anesthesia. A blocking experiment was performed for 64Cu-DOTA-VEGF121/rGel by injecting 200 μg of VEGF121 before injecting 64Cu-DOTA-VEGF121/rGel. 18F-FLT was synthesized by reacting no-carrier-added 18F-fluoride with the precursor 5′-O-benzoyl-2,3′-anhydrothymidine and then performing hydrolysis with 1% NaOH in a TRACERlab FXFN automatic synthesis module (GE Healthcare) (29). PET scans (3–20 min, static) were obtained at various time points up to 48 h after injection with 64Cu-DOTA-VEGF121/rGel and at 1 and 2 h after injection with 18F-FLT.
Histologic Analysis
Paraffin sections (7 μm thick) were deparaffinized in xylene, rehydrated in graded alcohols, transferred to PBS, and then stained with hematoxylin and eosin (H&E) (Biogenex Laboratories). For Ki67 staining, an antigen retrieval kit with pepsin was used in accordance with the manufacturer's guidelines (Abcam). Endogenous peroxidase was blocked with 3% hydrogen peroxide in PBS for 12 min. Sections were washed 3 times with PBS and incubated for 20 min at room temperature with a protein-blocking solution containing PBS (pH 7.5), 5% normal horse serum, and 1% normal goat serum. Excess blocking solution was drained, and the samples were incubated for 18 h at 4°C with a 1:100 dilution of Ki67 (Lab Vision). After being rinsed 4 times with PBS and incubated for 1 h at room temperature with the secondary antibody, the sections were rinsed with PBS and incubated for 5 min with diaminobenzidine (Research Genetics). Finally, the sections were attached to glass slides with mounting medium (Vectashield; Vector Labs), coverslips were added, and the sections were stored at −20°C.
Immunofluorescent Terminal Deoxynucleotidyltransferase-Mediated Biotin-dUTP Nick-End Labeling (TUNEL) Analysis
Paraffin sections were deparaffinized as described earlier. TUNEL analysis was performed by use of a commercial kit (Roche Applied Science) in accordance with the manufacturer's instructions. Samples were fixed with 10% formalin (methanol free) for 10 min at room temperature, washed with PBS, and permeabilized by incubation with 0.2% polyethylene glycol p-(1,1,3,3-tetramethylbutyl)-phenyl ether (Triton X-100; Sigma) in PBS (v/v) for 15 min. After the samples were incubated with equilibration buffer (from the kit), a reaction buffer containing equilibration buffer (45 mL), a nucleotide mixture (5 mL), and terminal deoxynucleotidyltransferase (1 mL) was added and incubated with the samples in a humidified chamber for 1 h at 37°C in the dark. The reaction was terminated by immersing the samples in 30 mM NaCl and 3 mM sodium citrate (pH 7.2) for 15 min; this step was followed by 3 washes to remove unincorporated fluorescein-dUTP. The nuclei were stained with 4′,6′-diamidino-2-phenylindole (DAPI; 1 mg/mL) for 10 min.
Immunofluorescence Staining
Frozen tissue sections (7 μm thick) were fixed with cold acetone. Samples were washed 3 times with PBS and incubated for 20 min at room temperature with a protein-blocking solution containing PBS (pH 7.5), 5% normal horse serum, and 1% normal goat serum. Excess blocking solution was drained, and the samples were incubated for 18 h at 4°C with a 1:100 dilution of rat monoclonal anti-CD31 antibody (BD PharMingen). Samples were rinsed 4 times with PBS and mounted with DAPI mounting medium for staining of the nuclei (Vectashield).
Fluorescence images were acquired by use of an Axiovert 200M fluorescence microscope (Carl Zeiss MicroImaging, Inc.) equipped with a DAPI (excitation 365 nm; emission 397 nm), Texas Red (excitation 535 nm; emission 610 nm), and Cy5.5 (excitation 665 nm; emission 725 nm) filter set. Images were obtained with a thermoelectrically cooled charge-coupled device (Micromax, model RTE/CCD-576; Princeton Instruments Inc.) and analyzed with MetaMorph software (version 6.2r4; Molecular Devices Corp.).
Data Processing and Statistics
All of the data presented are given as the mean ± SD of n independent measurements. Statistical analysis was performed with a 1-way ANOVA for multiple groups and an unpaired Student t test; statistical significance was assigned for P values of <0.05 (GraphPad Prism).
RESULTS
Binding and Functional Assays for DOTA-VEGF121/rGel
VEGF121/rGel and DOTA-VEGF121/rGel were able to inhibit 125I-VEGF165 binding to VEGFR-2 expressed on PAE/KDR cells in a dose-dependent manner (Fig. 1A). IC50 values of 24.5 and 40.6 nM were obtained for VEGF121/rGel and DOTA-VEGF121/rGel, respectively, indicating that no significant change in VEGF121/rGel binding affinity was caused by DOTA conjugation. Western blot analysis (functional assay) of VEGF121/rGel and DOTA-VEGF121/rGel with PAE/KDR cells revealed a slight decrease in the level of expression of phosphorylated KDR after DOTA conjugation (Fig. 1B). Increased levels of expression of phosphorylated KDR were observed at VEGF121/rGel and DOTA-VEGF121/rGel concentrations of ≥5 nM, with a consistent single protein band at 200 kDa.
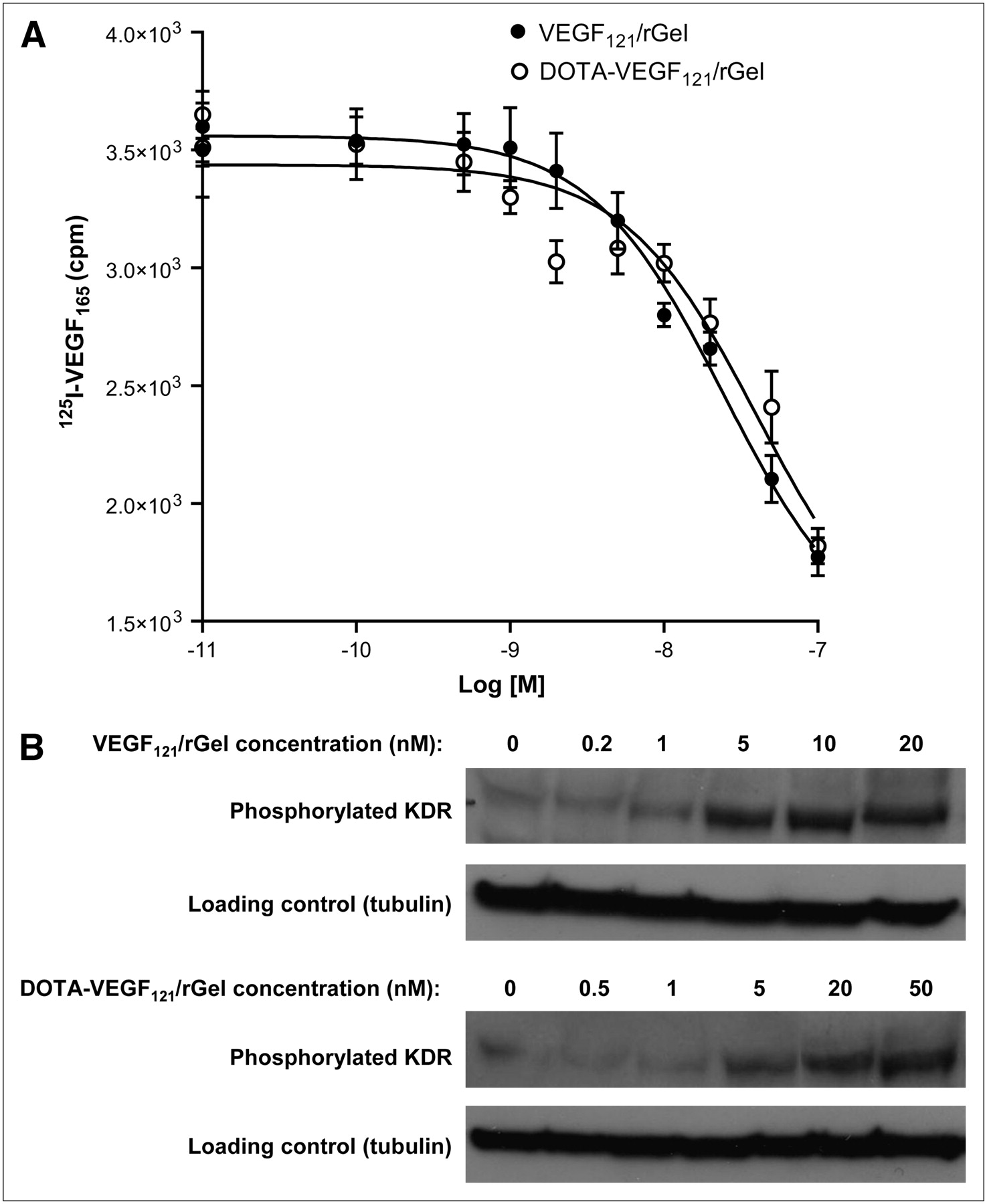
(A) Cell-binding assay revealed IC50 values of 24.5 and 40.6 nM for VEGF121/rGel and DOTA-VEGF121/rGel, respectively, demonstrating no significant difference in inhibition of 125I-VEGF165 binding to PAE/KDR cells after DOTA conjugation. (B) Functional assay revealed no significant difference in levels of phosphorylated KDR induced by serial concentrations of VEGF121/rGel and DOTA-VEGF121/rGel.
64Cu-DOTA-VEGF121/rGel Tumor Accumulation
64Cu labeling of DOTA-VEGF121/rGel, including final purification, took 90 ± 10 min (n = 3), and the radiolabeling yield was 85.2% ± 9.2% (on the basis of 37 MBq of 64Cu per 25 μg of DOTA-VEGF121/rGel; n = 3). The specific activity of 64Cu-DOTA-VEGF121/rGel was 1.3 ± 0.1 GBq/mg, and the radiochemical purity was greater than 98%. The number of DOTA molecules per VEGF121/rGel molecule was found to be 3.3 ± 0.1 (n = 4).
64Cu-DOTA-VEGF121/rGel exhibited high pretreatment tumor accumulation and retention and high tumor-to-background contrast from 1 to 48 h after injection in glioblastoma xenografts (Fig. 2A). Tumor accumulation at 1 h after injection was 5.8 ± 0.5 percentage injected dose per gram (%ID/g) (n = 3) and steadily increased and peaked at about 18 h after injection (11.8 ± 2.3 %ID/g). At 46 h after injection, tumor uptake decreased to 8.4 ± 1.7 %ID/g. Although the tumor uptake of 64Cu-DOTA-VEGF121/rGel remained high as tumor sizes increased, there was no clear relationship between tumor size and tracer uptake. 64Cu-DOTA-VEGF121/rGel was cleared through both the hepatic and the renal pathways (30). A blocking experiment with 200 μg of VEGF121 injected before 64Cu-DOTA-VEGF121/rGel injection revealed a significant reduction in 64Cu-DOTA-VEGF121/rGel uptake (Fig. 2B). PET clearly demonstrated VEGFR-specific tumor uptake of 64Cu-DOTA-VEGF121/rGel, a finding that provided the basis for the following treatment protocol. On the basis of the in vivo pharmacokinetics of 64Cu-DOTA-VEGF121/rGel, VEGF121/rGel was administered every other day for the VEGFR-2–targeted treatment of orthotopic U87MG glioblastomas.
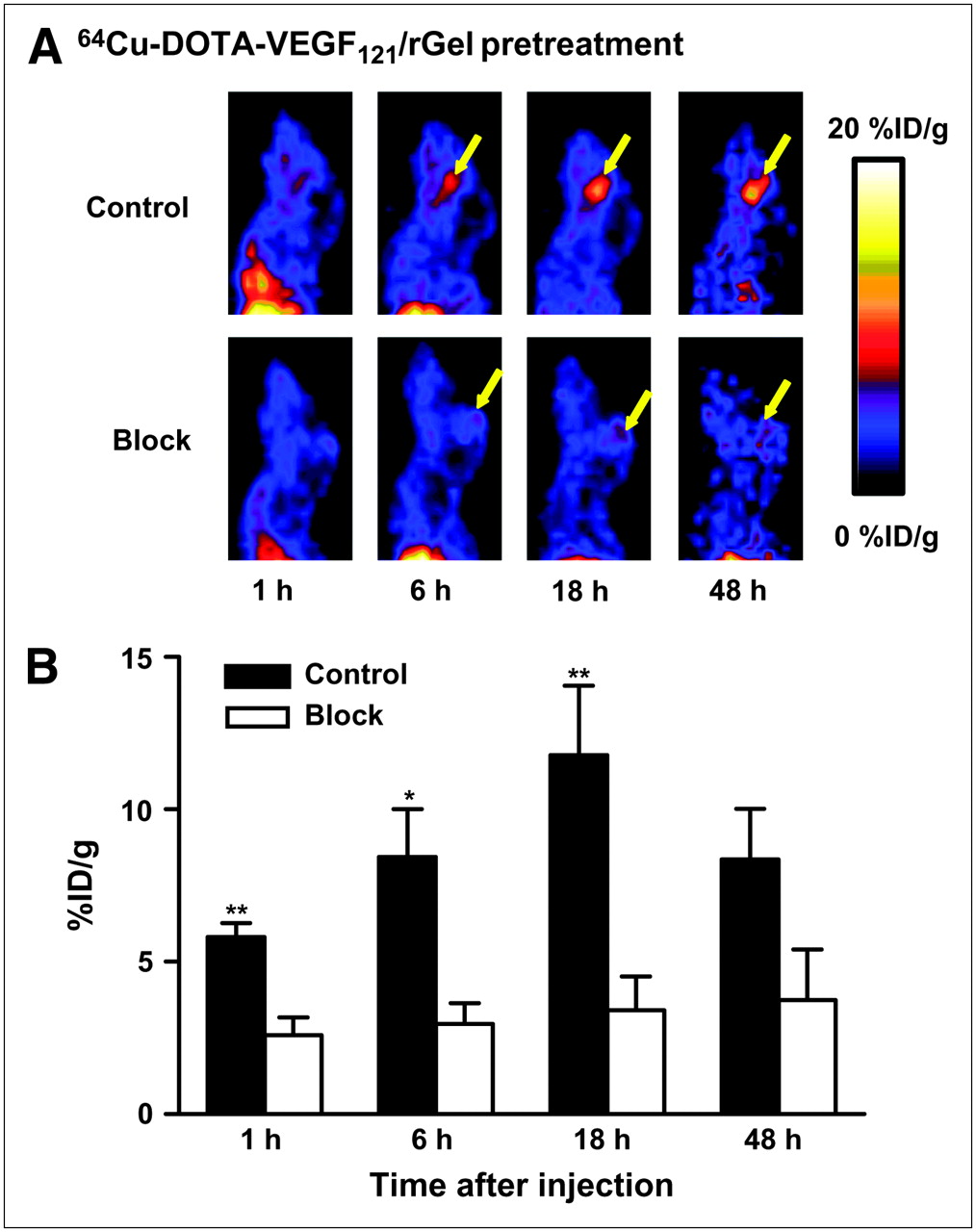
(A) Representative sagittal PET images of 64Cu-DOTA-VEGF121/rGel tumor accumulation from 1 h to 48 h after injection in glioblastoma tumor–bearing mouse (control). Tumor accumulation steadily increased and peaked at 11.8 ± 2.3 %ID/g (n = 3) at 18 h after injection. Sagittal PET images of tumor-bearing mouse injected with 200 μg of VEGF121 before 64Cu-DOTA-VEGF121/rGel injection are also shown (block). Yellow arrows indicate tumor location. (B) Blocking experiment with 200 μg of VEGF121 injected before 64Cu-DOTA-VEGF121/rGel injection revealed significant reduction in 64Cu-DOTA-VEGF121/rGel tumor uptake. *P < 0.05; **P < 0.01.
BLI and MRI Monitoring of Glioblastoma Growth Inhibition
Before treatment with VEGF121/rGel, we first validated the use of BLI for the assessment of longitudinal tumor growth in comparison with gadolinium-enhanced MRI. There was a significant increase in both BLI and MRI tumor signal intensities as tumors grew from day 21 to day 46 after inoculation, and logarithmic transformation of tumor volumes assessed by BLI and MRI on the same day showed a strong linear correlation (r = 0.89, n = 14). The MRI tumor volume spanned 1–74 mm3, and increases in tumor volume had no significant effect on the time required to reach the peak BLI tumor signal intensity from day 1 to day 46 after inoculation.
There were no significant differences in MRI tumor volume or BLI tumor signal intensity between the control group and the VEGF121/rGel treatment group on day 34 after inoculation. Control and treatment group tumor volumes (as determined by MRI) at baseline were 2.8 ± 0.8 and 3.9 ± 0.7 mm3, respectively (Figs. 3A and 3B). Peak BLI tumor signal intensities in the control and treatment groups were 2.9 × 107 ± 1.6 × 107 and 2.7 × 107 ± 1.3 × 107 photons per second, respectively (Figs. 3C and 3D). There were significant decreases in MRI tumor volume and BLI tumor signal intensities in the treatment group compared with the control group after only 2 of the 4 total doses of VEGF121/rGel. The MRI tumor volume in the control group increased to 6.3 ± 2.0 mm3, whereas that in the treatment group decreased to 2.9 ± 0.6 mm3 (P < 0.05). Similarly, the BLI tumor signal intensity in the control group increased to 7.0 × 107 ± 2.3 × 107 photons per second, whereas that in the treatment group increased only slightly, to 3.7 × 107 ± 1.2 × 107 photons per second (P < 0.05).

(A) MRI revealed no difference in tumor volume between groups (control: n = 6; treatment: n = 9) at baseline and decrease in treatment group tumor volume after 2 doses of VEGF121/rGel. Yellow arrows indicate tumor location. (B) From baseline to after 2 doses of VEGF121/rGel, control group tumor volume increased from 2.8 ± 0.8 mm3 to 6.3 ± 2.0 mm3, whereas treatment group tumor volume decreased 1.3-fold, from 3.9 ± 0.7 mm3 to 2.9 ± 0.6 mm3. *P < 0.05. (C) BLI of control and treatment groups revealed no difference in tumor signal intensity at baseline and significant difference after 2 doses of VEGF121/rGel. (D) Control group BLI tumor signal intensity increased from 2.9 × 107 ± 1.6 × 107 photons per second to 7.0 × 107 ± 2.3 × 107 photons per second from baseline to after 2 doses of VEGF121/rGel, whereas treatment group BLI tumor signal intensity increased only slightly, from 2.7 × 107 ± 1.3 × 107 photons per second to 3.7 × 107 ± 1.2 × 107 photons per second. *P < 0.05.
The difference in MRI tumor size between the 2 groups reached a peak after 4 doses of VEGF121/rGel, with a significant 8.7-fold-lower BLI tumor signal intensity in the treatment group than in the control group (P < 0.05) (Fig. 4). There was no difference in starting body weight between the 2 groups (control: 23.2 ± 0.5 g; treatment: 21.6 ± 0.5 g), but there was a significant difference in body weight loss between the 2 groups after therapy completion (control: 3.7 ± 0.3 g; treatment: 5.8 ± 0.7 g), possibly indicating some normal tissue cytotoxic side effects.

BLI tumor signal intensities for control and treatment groups from baseline to after 4 doses of VEGF121/rGel revealed a peak 8.7-fold decrease in BLI tumor signal intensity in treated mice. *P < 0.05.
18F-FLT PET Imaging of VEGF121/rGel Treatment
18F-FLT PET revealed significant differences in tumor uptake and the ratio of tumor uptake to background uptake (T/B ratio) between the control group and the treatment group (Fig. 5A), with no significant differences between 1 h and 2 h after injection. 18F-FLT tumor accumulation values at 1 h after injection (n = 3) were 2.9 ± 0.7 %ID/g in the control group and 1.7 ± 0.4 %ID/g after 4 doses of VEGF121/rGel (P < 0.05), indicating decreased DNA synthesis during treatment (Fig. 5B). Showing a trend similar to that for tumor uptake, the T/B ratios at 1 h after injection (n = 3) were 4.1 ± 0.7 and 2.3 ± 0.5 for the control and treatment groups, respectively (P < 0.05) (Fig. 5C). Both tumor uptake and the T/B ratio at 2 h after injection showed the same trends as those observed at 1 h after injection.

(A) Representative coronal and sagittal images of 18F-FLT at 1 h after injection for mouse before and after 4 doses of VEGF121/rGel treatment. Yellow arrows indicate tumor location. (B) Tumor accumulation values at 1 h after injection (n = 3) were 2.9 ± 0.7 %ID/g for control tumors and 1.7 ± 0.4 %ID/g for tumors after 4 doses of VEGF121/rGel. Quantification data from 18F-FLT PET scan at 2 h after injection are also shown. (C) T/B ratios at 1 h after injection were 4.1 ± 0.7 and 2.3 ± 0.5 for control and treated mice, respectively (n = 3). *P < 0.05. Data from 18F-FLT PET scan at 2 h after injection are similar to those at 1 h after injection.
Histologic Analysis of VEGF121/rGel Glioblastoma Inhibition
Histologic analysis of paraffin-embedded tumor sections stained with H&E, Ki67, and TUNEL revealed significant morphologic, proliferative, and apoptotic differences between the control group and the treatment group (Fig. 6A). In VEGF121/rGel-treated mice, H&E analysis revealed marked degradation and weakening of tumor neovasculature. In contrast, control mice showed healthy, mature tumor endothelium. Ki67 analysis revealed a 5 ± 2% proliferative index in the treated mice, whereas the control mice had a proliferative index of 17 ± 4%. Quantitative TUNEL-positive cell analysis revealed increased DNA fragmentation and apoptosis in the treated mice compared with the control mice. Tumors in the control mice had 4.1 ± 0.9% TUNEL-positive cells, whereas those in the treated mice had 12.8 ± 2.2% TUNEL-positive cells. Immunohistochemical analysis with DAPI, CD31, and TUNEL staining revealed increased apoptosis on tumor vasculature and surrounding cells in the treated mice compared with the control mice, indicating specific VEGF121/rGel-induced tumor vasculature damage (Fig. 6B). These ex vivo results validated our initial findings from 18F-FLT PET and supported our hypothesis that VEGF121/rGel inhibits glioblastoma growth through tumor vasculature degradation and subsequent decreases in DNA synthesis and increases in tumor cell apoptosis.

(A) Ex vivo histologic and immunohistochemical analyses with H&E, Ki67, and TUNEL at ×20 magnification revealed clear differences between control tumors and tumors after 4 doses of VEGF121/rGel. H&E analysis revealed VEGF121/rGel-induced damage of tumor vasculature accompanied by inflammation and red blood cell extravasation. Ki67 analysis revealed significant decreases in DNA synthesis and proliferative index in treated tumors compared with control tumors (brown cells are Ki67-positive cells). Quantitative TUNEL analysis demonstrated marked increases in DNA fragmentation and apoptosis in treated mice compared with control mice (green cells are TUNEL-positive cells). (B) Immunofluorescence analysis with DAPI (blue), CD31 (green), and TUNEL (red) at ×10 magnification revealed significant increases in apoptosis on tumor vasculature and surrounding cells in treated mice compared with control mice, indicating specific VEGF121/rGel-induced tumor vasculature damage.
DISCUSSION
We have demonstrated the use of multimodality molecular imaging to monitor VEGF121/rGel inhibition of orthotopic glioblastoma growth through specific tumor vasculature degradation. We have also shown that noninvasive BLI, MRI, and PET can effectively track and quantify the treatment response, and we have verified a strong correlation between BLI and gadolinium-enhanced MRI for intracranial tumor growth surveillance. PET with 64Cu-DOTA-VEGF121/rGel revealed high fusion toxin tumor accumulation before treatment, validating the tumor-targeting efficacy and elucidating the pharmacokinetics of the fusion toxin in vivo; on the basis of these findings, the dose interval of every other day was chosen. 18F-FLT imaging reflected subsequent VEGF121/rGel-induced decreases in tumor DNA synthesis, a finding that was verified by histologic analysis. Similar to previous results (8,15,17), histologic analysis revealed specific damage to tumor vascular endothelium and not tumor cells themselves, indicating that the antitumor effects of VEGF121/rGel were attributable to antiangiogenic mechanisms and secondary hypoxia-induced cell death rather than direct tumor cell cytotoxicity.
GBM is characterized by rapid cellular proliferation, which leads to hypoxic conditions within the tumor; these conditions can then trigger angiogenesis by releasing humoral factors to recruit new blood supplies (31). Genes encoding VEGF and VEGFR-2 have been found to be amplified in primary glioblastomas and may also be upregulated in secondary glioblastomas (32). The blood–brain barrier (BBB) is mediated by endothelial tight junctions and is defective in glioblastomas. This dysfunction results in the characteristic cerebral edema and contrast enhancement on MRI that accompany glioblastomas. The mechanisms underlying the breakdown of the BBB caused by glioblastomas are essentially unknown. Because nonneoplastic astrocytes are required to induce the BBB features of cerebral endothelial cells, it is possible that malignant astrocytes in glioblastomas have lost this ability because of dedifferentiation. Glioma cells may also actively degrade previously intact BBB tight junctions (33). The disrupted BBB facilitates the effective delivery of high-molecular-weight probes, such as VEGF121/rGel, to the intracranial glioblastoma tumor.
Key findings of the present study were high tumor accumulation and a long duration of binding (>48 h) of 64Cu-DOTA-VEGF121/rGel, indicating favorable pharmacokinetics for nuclear imaging and therapy. 64Cu-DOTA-VEGF121/rGel demonstrated faster uptake and clearance than higher-molecular-weight anti-VEGF antibodies, which take longer to accumulate in tumors and clear from the circulation, often causing a sustained background signal and prolonged immunogenicity (30). The main disadvantage of VEGFR-targeted therapies is that protein binding to different members of the VEGFR superfamily may also affect systems other than angiogenesis. 64Cu-DOTA-VEGF121/rGel showed prominent uptake in the kidneys (10–20 %ID/g; data not shown). Although VEGF121/rGel exerts high toxicity specifically for VEGFR-2 and not VEGFR-1 (15), the toxic side effects observed in the present study may be partly attributed to the high level of VEGFR-1 expression in the kidneys.
Previous studies showed no significant in vivo cytotoxic effects of VEGF121/rGel in normal tissues, as KDR is likely expressed at insufficient levels to cause significant VEGF121/rGel binding and vascular damage in normal organs, such as the kidney glomerulus (8,15,17,34). In addition, the total number of VEGFRs on normal organ vascular endothelium is significantly lower than that on tumor vasculature (34), and it was previously shown that a threshold number of KDR receptors is needed (∼2 × 103 receptors per cell) before specific VEGF121/rGel cytotoxic effects are observed (15). Toxicology studies with VEGF121/rGel in mice indicated no specific renal or hepatic toxicity, even at the MTD, with no elevations in the levels of hepatic enzymes as evidence of functional organ damage (17).
Several challenges need to be overcome to ensure successful clinical translation of antiangiogenic therapies; the greatest of these are temporal dependence and insufficient antitumor effects. It has been hypothesized that antiangiogenic therapies will be most effective against small tumors and should be administered before the development of a well-established vascular network for optimal outcomes (35). Previous work with VEGF121/rGel and pulmonary breast tumor metastases revealed greater tumor vasculature susceptibility to treatment in small metastatic lesions than in larger metastases (diameters of >500 μm) (17). This finding was likely attributable to a higher VEGFR density on a small, rapidly expanding tumor vasculature; decreased recruitment of supporting cells, such as pericytes and smooth muscle; differences in basement membrane integrity; or a combination of these factors (17). Therefore, imaging guidance is critical to VEGFR-targeted therapies, as the therapeutic window is usually quite narrow. PET of VEGFR expression can play a very important role in determining whether and when to start VEGFR-targeted cancer therapy, as it can provide a straightforward and convenient way to monitor VEGFR expression levels in vivo.
In recent years, in vivo PET of VEGF radioligands has shown promise as a means for effectively monitoring tumor angiogenesis. 124I-labeled monoclonal antibody 124I-SHPP-VG76e has shown high tumor-to-background contrast in a human fibrosarcoma model (36), and PET has been demonstrated to be successful in glioblastoma xenografts with 111In-hnTf-VEGF165 (37). We recently reported the use of 64Cu-labeled VEGF121 for PET of tumor VEGFR expression (22). Small-animal PET imaging revealed rapid and high 64Cu-DOTA-VEGF121 uptake in tumors with a high level of VEGFR expression but very low and sporadic uptake in tumors with a low level of VEGFR expression. The future of anticancer imaging and therapy is in combinatorial approaches that simultaneously target and visualize tumor cells to reduce tumor burden as well as the corresponding vascular support system to prevent neovascularization, the growth of metastatic foci, and the formation of new metastatic lesions. Preclinical studies have shown significantly improved antitumor effects in vivo with vascular ablation therapies in combination with either radiotherapy (38) or chemotherapy (39). It is unlikely that antiangiogenic therapies acting alone within cytotoxicity limits will be able to completely inhibit tumor growth within the time frame available for treatment. Therefore, in the future, it will be critical to develop antiangiogenic therapies with increased binding specificity and tumor vasculature toxicity in addition to decreased antigenicity and size.
Preclinical data obtained with various tumor models have shown impressive antiangiogenic and antitumor effects with VEGF121/rGel (8,15,17), and our Investigational New Drug (IND) submission for a Phase I trial of VEGF121/rGel in patients with solid tumors is currently being approved by the U.S. Food and Drug Administration. In addition, clinical trial results obtained with an anti-CD33 HuM195/rGel fusion protein have shown low rGel antigenicity and no observed hepatotoxicity or incidences of vascular leak syndrome (40). The results of the present study verify the antitumor efficacy of VEGF121/rGel treatment in an orthotopic glioblastoma model while demonstrating the applicability of preclinical optical imaging and clinically relevant PET for monitoring treatment efficacy. At present, clinical PET is limited by a lack of radioisotopes (e.g., 64Cu), multifunctional probes, and cost-effectiveness. Therefore, it is imperative that markers that are increasingly tumor specific be identified and targeted to optimize clinical therapy. It will also be important in the future to investigate the possible additive benefit of antiangiogenic therapies with established radiotherapy and chemotherapy protocols while determining the molecular profiles of different tumors to aid in the design of optimal treatments through the use of proteomics.
CONCLUSION
We have shown VEGFR-2–targeted imaging and therapy with VEGF121/rGel to be an effective means for monitoring and treating orthotopic glioblastoma in a mouse model. On the basis of our results, we believe that future research on and clinical translation of VEGF121/rGel-based imaging and therapy are needed and may allow for the identification and stratification of patients who will benefit from fusion toxin therapy after tumor resection. Additional preclinical and clinical studies are warranted to optimize the imaging, timing, dosing, and therapeutic effects of this fusion protein alone and in combination with adjuvant therapies to improve patient prognosis and survival rates.
Acknowledgments
We thank the cyclotron team at the University of Wisconsin–Madison for 64Cu production. We also thank Drs. Fredrick T. Chin, Murugesan Subbarayan, and David W. Dick for the production of 18F-FLT. Funding for this study was provided by the National Institute of Biomedical Imaging and Bioengineering (NIBIB) (R21 EB001785), the National Cancer Institute (NCI) (R21 CA102123, P50 CA114747, U54 CA119367, and R24 CA93862), the Department of Defense (DOD) (W81XWH-04-1-0697, W81XWH-06-1-0665, W81XWH-06-1-0042, and DAMD17-03-1-0143), a Benedict Cassen Postdoctoral Fellowship from the Education and Research Foundation of the Society of Nuclear Medicine, an American Brain Tumor Association Fellowship, and a Stanford University School of Medicine Medical Scholars Award. Research conducted, in part, by the Clayton Foundation for Research.
Footnotes
-
↵* Contributed equally to this work.
References
- Received for publication October 9, 2006.
- Accepted for publication December 3, 2006.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Specific biomarkers of receptors, pathways of inhibition and targeted therapies: pre-clinical developments
- Inhibition of Prostate Cancer Osteoblastic Progression with VEGF121/rGel, a Single Agent Targeting Osteoblasts, Osteoclasts, and Tumor Neovasculature
- Multimodality Molecular Imaging of Tumor Angiogenesis
- Dynamic Small-Animal PET Imaging of Tumor Proliferation with 3'-Deoxy-3'-18F-Fluorothymidine in a Genetically Engineered Mouse Model of High-Grade Gliomas