


Abstract
In mice bearing human breast cancer xenografts, we examined the biodistribution of 18F-fluorocyclophosphamide (18F-F-CP) to evaluate its potential as a noninvasive prognostic tool for predicting the resistance of tumors to cyclophosphamide therapy. Methods: 18F-F-CP was synthesized as we recently described, and PET data were acquired after administration of 18F-F-CP in mice bearing human breast cancer xenografts (MCF-7 cells). Tracer biodistribution in reconstructed images was quantified by region-of-interest analysis. Distribution was also assessed by harvesting dissected organs, tumors, and blood, determining 18F content in each tissue with a γ-well counter. The mice were subsequently treated with cyclophosphamide, and tumor size was monitored for at least 3 wk after chemotherapy administration. Results: The distribution of harvested activity correlated strongly with distribution observed in PET images. Target organs were related to routes of metabolism and excretion. 18F-F-CP uptake was highest in kidneys, lowest in brain, and intermediate in tumors, as determined by both image-based and tissue-based measurements. 18F-F-CP uptake was not inhibited by coadministration of an approximately ×700 concentration of unlabeled cyclophosphamide. PET measures of 18F-F-CP uptake in tumor predicted the magnitude of the response to subsequent administration of cyclophosphamide. Conclusion: Noninvasive assessment of 18F-F-CP uptake using PET may potentially be helpful for predicting the response of breast tumors to cyclophosphamide before therapy begins.
- 18F
- cyclophosphamide
- prognostic imaging
- positron emission tomography
- breast tumor therapy
- breast cancer
- small-animal imaging
Cyclophosphamide is one of the most widely used agents in cancer chemotherapy. Its clinical utility has been demonstrated against a relatively broad spectrum of human cancers (1), including malignant lymphomas such as Hodgkin's disease (2,3), leukemias (4), neuroblastomas (5), and breast carcinomas (6), prompting numerous investigations regarding the metabolism, mechanism of action, and influence of structural modifications on the therapeutic efficacy of this drug. The metabolic fate of cyclophosphamide and its analogs is long-established (7). Enzyme-catalyzed oxidation of cyclophosphamide by cytochrome p450 produces 4-hydroxycyclophosphamide (8). The subsequent formation of aldophosphamide, with which it is in equilibrium, leads through a nonenzymatic step to cleavage into acrolein and phosphoramide mustard, the ultimate major alkylating species. However, it has been realized that individual differences in the metabolism of cyclophosphamide and its analogs (e.g., ifosfamide) can influence the clinical outcome of therapy (9,10).
Adverse effects of cyclophosphamide, including nausea, vomiting, diarrhea, leukopenia, renal insufficiency, hemorrhagic cystitis, and colitis, can be severe, and the ability to garner knowledge about its potential efficacy before treatment would be of substantial clinical value. Predicting resistance to cyclophosphamide is complicated and has been unreliable using in vitro methods alone. Several groups have shown that cancer cell lines in vitro quickly develop resistance to cyclophosphamide after incubation with the compound (11–13). In addition, the literature suggests that prior exposure to any alkylating agent increases the likelihood of resistance to cyclophosphamide (11–14). Both genetic and physiologic alterations can occur to render the cells resistant to cyclophosphamide (15–17), including the regulation of cellular levels of aldehyde dehydrogenase (10,18) and the activity of glutathione S-transferase (11). Information about the biodistribution and kinetics of the metabolism of cytotoxic drugs in each patient could improve the selection of appropriate chemotherapeutic regimens by assisting with individualization of therapy. Consequently, breast cancer patients might more often be spared the toxicities of chemotherapy regimens that have little or no value for them.
PET is a functional imaging tool that has been shown to have high accuracy in screening and staging many types of cancer (19–28). PET and SPECT (25,29–32) are also used to assess the ultimate response to chemotherapy in patients with breast cancer. Nuclear medicine imaging modalities could potentially be applied to study tracer levels of chemotherapeutics before therapy, allowing for a more rational approach to the selection of pharmacologic agents.
We recently reported the radiosynthesis of the PET analog of cyclophosphamide, [2-[(2-chloro-2′-18F-fluorodiethyl)amino]-2H-1,3,2-oxazaphosphorinane-2-oxide, or 18F-fluorocyclophosphamide (18F-F-CP) (33). In the present work, we examined the feasibility of using PET to noninvasively estimate uptake of cyclophosphamide in vivo, as a tool to predict response to subsequent treatment with cyclophosphamide.
MATERIALS AND METHODS
Materials
Unless otherwise noted, all chemicals were purchased from Sigma-Aldrich or Fisher Scientific. MCF-7 (American Type Culture Collection) cells were propagated in RPMI medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen), 2 mM L-glutamine (Invitrogen), and 1% penicillin/streptomycin (Invitrogen). All cell lines were maintained in a humidified 37°C/5% CO2 environment.
All animal studies were performed under a protocol approved by the Chancellor's Animal Research Committee of UCLA. MCF-7 cells, which form xenografts in female athymic mice, were injected subcutaneously at approximately 3.0 × 107 cells per tumor on the shoulder region of 4- to 6-wk-old female athymic mice weighing from 20 to 30 g (Charles River Laboratories). Before cell injection, all mice were primed with 17β-estradiol (Innovative Research of America) applied subcutaneously (1.7 mg of estradiol per pellet) to promote tumor growth. The tumors were measured frequently from the day of injection to the day of imaging. At the time of imaging (10–18 d after the time of injection), the tumors ranged from 273 to 1,364 mm3.
In Vitro 18F-F-CP Cytotoxicity Assay
The cytotoxic effect of 18F-F-CP was compared with that of cyclophosphamide and assayed using the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide)-based in vitro toxicology assay kit (Sigma-Aldrich). Briefly, MCF-7 cells were seeded in 24-well tissue culture plates (BD Falcon) at 105 cells per well in 0.5 mL of complete medium. The compounds were dissolved in saline and then diluted in complete fresh medium to various concentration levels. To each well, 0.5 mL of drug in medium was added. To obtain active metabolites of the compounds, we first activated each drug by incubation with human liver microsomes as follows: 0.5 mg of liver microsomes (In Vitro Technologies) was preincubated at 37°C with 1 mg of drug in a 50 mM potassium phosphate buffer, with a pH of 7.4 and a final volume of 750 μL, with shaking for 5 min. A 250-μL volume of NADPH regenerating system buffer (b-NADP [0.5 mg/mL], glucose-6-phosphate [2 mg/mL], glucose-6-phosphate dehydrogenase [1.5 units/mL] in 2% NaHCO3) was added to each tube and incubated at 37°C, with shaking. After 60 min, 1 mL of methanol was added to each tube, and the tubes were centrifuged for 5 min. The resultant supernatant was diluted in complete fresh medium to various concentrations, based on the initial concentration of drug in the activation mixture. To each well, 0.5 mL of drug/medium was added. For each concentration, 4 replicate wells were treated, and the plates were incubated for 24 h. On the day of an assay, the MTT-based in vitro toxicology assay kit was used per included directions. The absorbance was read at 570 nm (690-nm reference) on a Genios microplate reader (Tecan).
Biodistribution and Organ Harvesting
For biodistribution studies, 28 healthy nude mice or nude mice bearing MCF-7 tumor xenografts were injected intraperitoneally with a solution of sterile saline containing approximately 9.25 MBq of 18F-F-CP prepared as previously described (33). Fifteen of these mice also were studied with small-animal imaging for comparison of distribution assessments. The animals were sacrificed by an injection of intracardiac pentobarbital (Nembutal [Abbott Laboratories]; 100 mg/kg of body weight) after an uptake time of 1 h. After sacrifice, all visceral organs and blood were collected from each animal. The organs were weighed, and γ-counting was performed to assess the amount of 18F radioactivity in each organ. Data were expressed as percentage injected dose per gram.
Tumor volumes for these studies and the predictive studies were estimated by an area measurement of length × width (mm2), with volumes calculated as (√area)3. These volumes averaged 630 ± 310 mm3 (mean ± SD) and ranged from 270 to 1,400 mm3 at the time of harvesting in biodistribution studies or of initial treatment in prediction studies.
Small-Animal PET and Predictive Value of 18F-F-CP
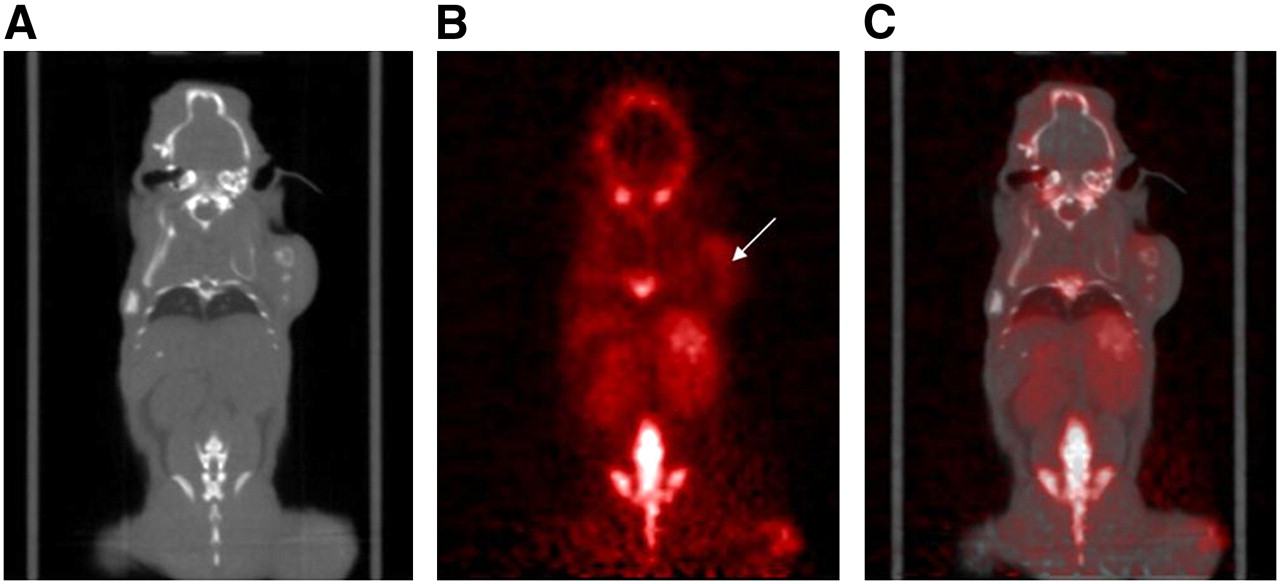
Forty minutes before small-animal PET scans, the mice were anesthetized by inhalation of 2% isoflurane and injected intraperitoneally with approximately 9.25 MBq of 18F-F-CP and varying amounts of unlabeled cyclophosphamide (Acros Organics USA), based on individual animal weights determined immediately before injection (0–5 mg/kg). Whole-body scans were acquired for 15 mice over 5 min in 1 bed position using a P4 microPET scanner (Concorde Microsystems Inc.). Some animals were injected with the tracer solution and immediately imaged to obtain a complete dynamic sequence over 30, 60, or 90 min. The mice were set prone in the scanner, with the long axis of the mouse parallel to the long axis of the scanner detectors. All mice were sacrificed immediately after PET data acquisition by an overdose of pentobarbital administered intracardially, and organs were harvested as described in the previous section to obtain biodistribution data corresponding to PET image data. A small-animal CT scan (MicroCAT II; ImTrek) was obtained for illustrative purposes for Figure 4 but was not routinely used in the cyclophosphamide in vivo biodistribution studies.
For predictive studies, 13 additional animals underwent the same PET procedures as described above and were imaged with small-animal CT for 10 min immediately after the PET scans. Images were then fused to capture the tumor location for tracer region-of-interest analysis. Immediately after imaging, the mice were treated intraperitoneally with a single dose of cyclophosphamide (100 mg/kg) in saline. Treatment efficacy was monitored for at least 3 wk. All tumors were monitored by serial micrometer measurements at least once a week.
All PET images were reconstructed by filtered backprojection with a matrix size of 128 × 128 × 63, a slice thickness of 1.21 mm, and a voxel size of 0.174 mm3 and were analyzed by a standard region-of-interest method using AMIDE (UCLA-Pharmacology) software, with quantification of decay-corrected regional activity normalized to injected dose concentration, in standardized uptake value units.
RESULTS
Comparison of In Vitro Toxicities of Fluorocyclophosphamide and Cyclophosphamide
Although the toxic activity of unlabeled F-CP was assayed in several other cancer cell lines by Papanastassiou et al. (34), they did not establish the relative toxicity of this compound to human breast cancers. We compared the cytotoxicity of F-CP to that of cyclophosphamide in MCF-7 human breast cancers in vitro. The toxicity of cyclophosphamide and of F-CP activated by cytochrome p450 was also compared with that of the compounds without enzymatic treatment (Fig. 1). For both cyclophosphamide and 18F-F-CP, the activated compounds were more toxic than the nonactivated compounds. At the lowest dose given, 0.0125 mg of drug per milliliter, the enzymatically activated forms of the drugs reduced the number of viable cells by 35%–40%. A more than 20-fold increase in concentration was required to achieve the same level of toxicity with drugs that were not metabolically activated. At the highest concentration (0.25 mg/mL), both 18F-F-CP and cyclophosphamide were significantly more toxic to MCF-7 human breast cancer cells after activation than were the original compounds before their activation (P < 0.01, Student t test).
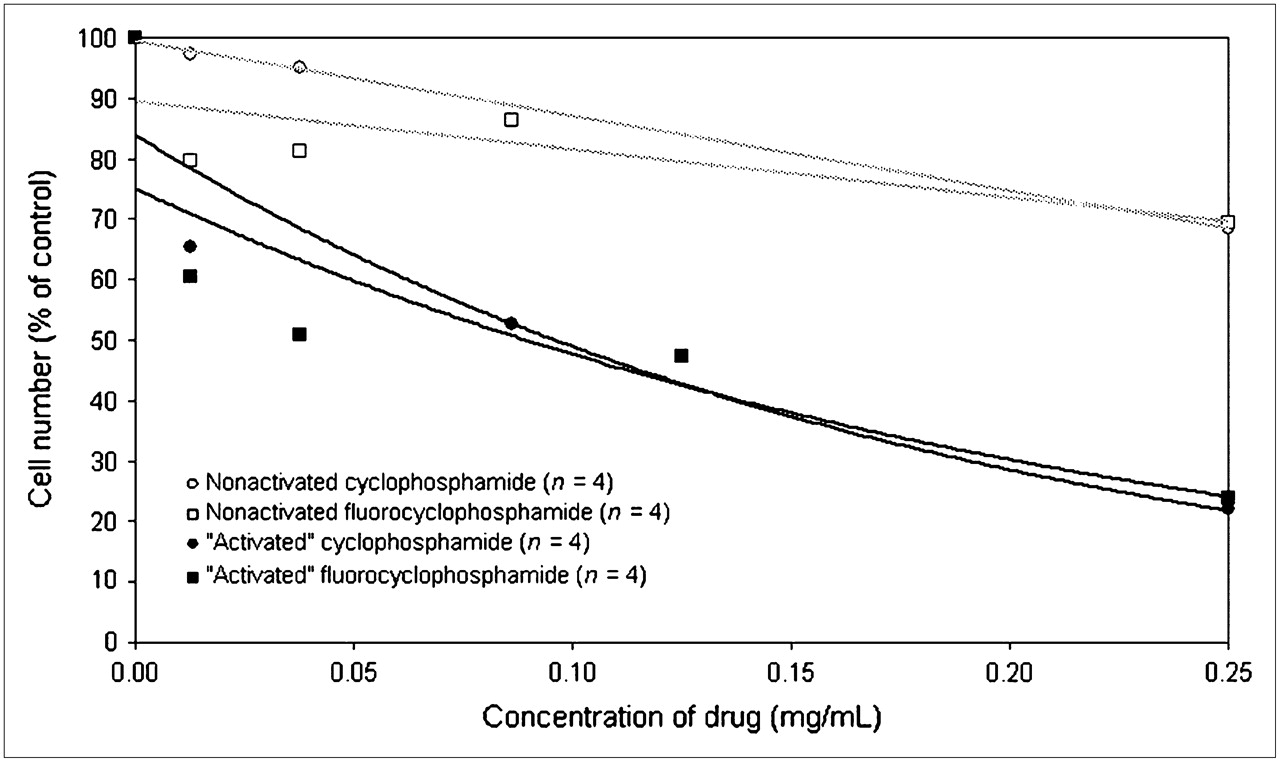
Comparison of relative cytotoxicity of cyclophosphamide and fluorocyclophosphamide in MCF-7 human breast cancer cells. Data are number of living cells after treatment at increasing doses, expressed as percentage of control (0 mg of drug per milliliter). “Activated” indicates drug that was preincubated with human liver microsomes before addition to cells.
Selection of Administration Route, Tracer Vehicle, and Uptake Time
Cyclophosphamide is generally administered to mice by intraperitoneal injection (35). In humans, cyclophosphamide is administered successfully by a variety of methods. In a test of the compatibility of intraperitoneal injections and small-animal PET imaging, the mice underwent dynamic PET up to 90 min after intraperitoneal injection of 18F-F-CP. 18F-F-CP was quickly mobilized from the peritoneal cavity into blood and organs, thus enabling scanning with minimal distress to the mice.
Because 18F-F-CP is a novel tracer, no previous data were available to determine the uptake time for imaging purposes. We therefore studied the time course of 18F-F-CP to determine the uptake period that optimized the tumor-to-background ratio. We found that the pharmacokinetics of 18F-F-CP uptake in the tumor were linear after 40 min and were stable in relation to other organs, in mice examined for more than twice that time (Fig. 2). A standard uptake time of 40 min for 18F-F-CP was thus selected for further biodistribution and imaging purposes.

Uptake of 18F-F-CP in MCF-7 tumor of nude mouse becomes approximately linear after 30 min. Line indicates best linear fit for 30–90 min.
18F-F-CP Behavior in the Presence of Pharmacologic Doses of Unlabeled Cyclophosphamide
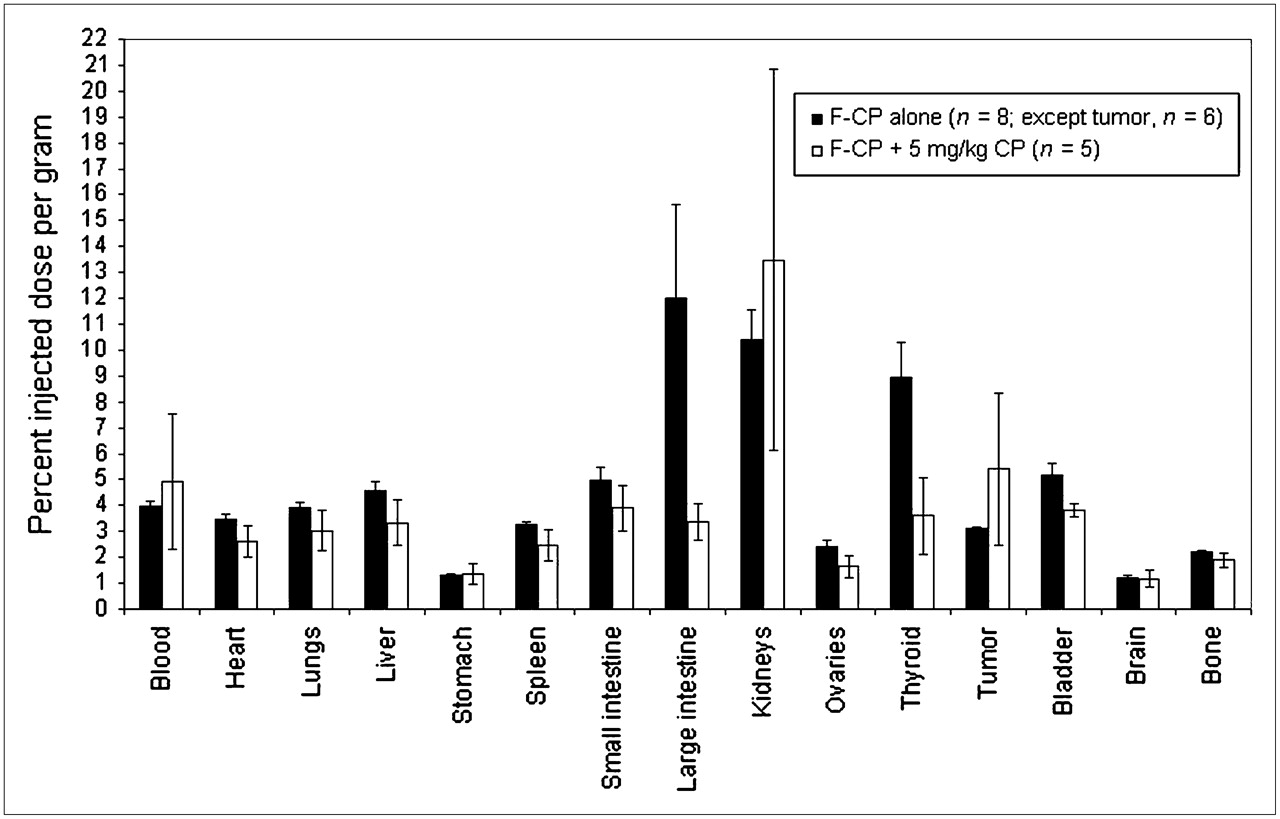
Pharmacokinetic and pharmacodynamic behavior can differ between drugs at pharmacologic doses and drugs at tracer doses. Because the amount of tracer injected for imaging can be 1,000 times less than the pharmacologic dose of unlabeled agent, it was important to test whether the behavior of the tracer was altered by the overall dose present. Therefore, the effect of increasing amounts of unlabeled cyclophosphamide on the biodistribution of 18F-F-CP in female nude mice bearing MCF-7 tumors was investigated (Fig. 3). (A concentration of ×1 is equivalent to 5 mg of CP per kilogram of the animal's weight.) The concentrations tested included ×0, ×0.03, ×0.1, ×0.3, and ×1. Increasing the amounts of cold cyclophosphamide did not appreciably change the overall biodistribution of 18F-F-CP to any organs investigated. Uptake measurements made by imaging the tracer would thus not be expected to overestimate the amount of cyclophosphamide available to the organ when administered at pharmacologic doses, as could occur if uptake proceeded by a process that was saturable in this dose range.

Effect of coadministration of pharmacologic dose of unlabeled cyclophosphamide on relative uptake of 18F-F-CP as measured by organ harvesting after 1 h of uptake.
Correlation of PET-Determined Uptake with Harvested Uptake and Subsequent Tumor Response
Uptake of 18F activity as assessed by PET (Fig. 4) after intraperitoneal injection of 18F-F-CP corresponded to that measured by organ harvesting (Fig. 5). The high correspondence between uptake measured by PET and that determined by direct counting of tissue-associated radioactivity (Pearson correlation coefficient, 0.957; P < 0.0001; n = 15; Student t test) suggested that 18F-F-CP imaging data could be used to assess the availability of tracer to tumor and other tissues in mice. In assessing the predictive value of 18F-F-CP in mice with breast cancer xenografts, we found a significant correlation between initial tumoral 18F-F-CP uptake and response to cyclophosphamide treatment with respect to regression of tumor. The tumor with the highest initial uptake of 18F-F-CP (standardized uptake value, 1.2) before treatment was the most responsive to treatment, having a 78% decrease in volume, whereas the tumor with the least uptake of 18F-F-CP (standardized uptake value, 0.3) demonstrated chemotherapy resistance (3% increase in volume). Tumors with intermediate levels of uptake tended to have intermediate responses to therapy (Fig. 6; n = 13, P = 0.01, r = 0.61).

CT (A), PET (B), and whole-body fused PET/CT (C) image of MCF-7 tumor, subcutaneously injected on right shoulder. Images represent tomographic coronal slices of tumor (white arrow) imaged 40 min after intraperitoneal injection of ∼9 MBq of 18F-F-CP. Animals were imaged prone such that left side of image corresponds to left side of animal.

Correlation of 18F activity assessed by PET (standardized uptake value) and by tissue harvesting (percentage injected dose per gram) after administration of 18F-F-CP.
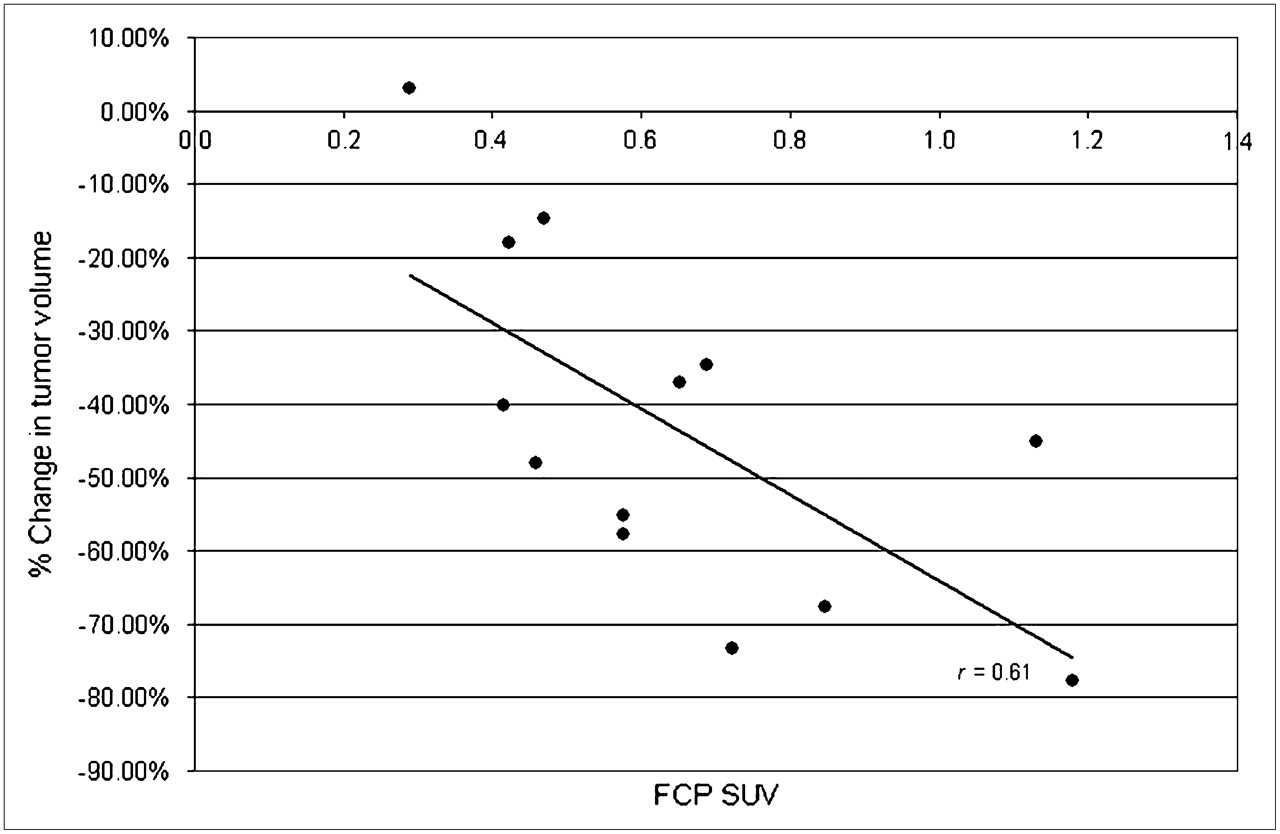
Correlation of initial 18F-F-CP standardized uptake value (SUV) and subsequent change in tumor volume after 21 d of chemotherapy in female athymic mice bearing MCF-7 tumors (n = 13; P = 0.01; r = 0.61; data pooled from 3 independent experiments).
DISCUSSION
Structure-activity studies of halogenated cyclophosphamide analogs suggest that certain modifications of the native compound do not significantly affect the metabolism and antitumor activity of the drug (36–39). On the basis of these studies, it was reasonable to suspect that the 18F-F-CP compound synthesized here would possess tumor uptake properties similar to those of cyclophosphamide. Cyclophosphamide is a prodrug that requires metabolism by cytochrome p450 to produce its toxic effects (8). The striking change in the toxicity of F-CP administered to cells before and after cytochrome p450 activation suggests that it, like cyclophosphamide, is a similar substrate for p450 metabolism. The biodistribution of F-CP does not vary greatly from that of cyclophosphamide, and both compounds are taken up extensively by the liver, bladder, and kidneys, organs involved in the metabolism and excretion of cyclophosphamide. Tracer uptake was not inhibited in the presence of pharmacologic doses of cyclophosphamide, indicating that measurements made by imaging tracer uptake would not overestimate the amount of cyclophosphamide that would be available to the tumor when administered at pharmacologic doses, and the correlation between tracer uptake assessed in harvested organs and by imaging methods was high. Those data suggested that 18F-F-CP PET could serve as an appropriate model with which to predict cyclophosphamide behavior in vivo. The tumor response studies further supported the potential utility of 18F-F-CP as a tool to predict the chemotherapeutic response to cyclophosphamide, with the responsiveness to chemotherapy correlating with the magnitude of tracer uptake in tumor before treatment.
CONCLUSION
This study, which provided the first (to our knowledge) PET images obtained with the radiofluorinated tracer analog of the commonly used chemotherapeutic agent cyclophosphamide, showed 18F-F-CP mainly in organs that relate to the metabolism and excretion of cyclophosphamide (hepatic and urinary), a result consistent with biodistribution data from organ harvesting. Uptake of 18F-F-CP in tumor was not high relative to that in other soft tissues, and 18F-F-CP could not serve as a substitute for 18F-FDG or other tracers as a tool to detect breast cancers. 18F-F-CP did, however, reach tumors in measurable amounts, and quantifying its uptake with PET could potentially be useful in the assessment of possible physiologic resistance in vivo, as indicated by the biodistribution and predictive studies reported here.
Acknowledgments
We thank Dr. Nagichettiar Satyamurthy, Dr. Arion Chatziioannou, Dr. David Stout, Dr. Waldemar Ladno, and Judy Edwards for their professional contributions related to image acquisition. Special thanks go to Katelyn McCabe and Melehia Frauenholtz for their roles in performing experiments, as well as to Dr. Anne Gangloff for useful discussions. We also acknowledge the help and support of the staff of the Ahmanson Biological Imaging Center. One of the authors is supported by a grant from NIH Research Training in Pharmacological Sciences and by the University of California Research Mentorship Program. This work was supported by California Breast Cancer Research Program grant 7KB0155.
Footnotes
-
Guest Editor: Kenneth A. Krohn, University of Washington Medical Center
-
COPYRIGHT © 2007 by the Society of Nuclear Medicine, Inc.
References
- Received for publication January 1, 2007.
- Accepted for publication August 27, 2007.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}