


Abstract
The norepinephrine analog 11C-meta-hydroxyephedrine (HED) is used with PET to map the regional distribution of cardiac sympathetic neurons. HED is rapidly transported into sympathetic neurons by the norepinephrine transporter (NET) and stored in vesicles. Although much is known about the neuronal mechanisms of HED uptake and retention, there is little information about the functional relationship between HED retention and cardiac sympathetic nerve density. The goal of this study was to characterize the dependence of HED retention on nerve density in rats with graded levels of cardiac denervation induced chemically with the neurotoxin 6-hydroxydopamine (6-OHDA). Methods: Thirty male Sprague–Dawley rats were divided into 6 groups, and each group was administered a different dose of 6-OHDA: 0 (controls), 7, 11, 15, 22, and 100 mg/kg intraperitoneally. One day after 6-OHDA injection, HED (3.7–8.3 MBq) was injected intravenously into each animal and HED concentrations in heart and blood at 30 min after injection were determined. Heart tissues were frozen and later processed by tissue homogenization and differential centrifugation into a membrane preparation for in vitro measurement of cardiac NET density. A saturation binding assay using 3H-mazindol as the radioligand was used to measure NET density (maximum number of binding sites [Bmax], fmol/mg protein) for each heart. Results: In control animals, NET Bmax was 388 ± 23 fmol/mg protein and HED heart uptake (HU) at 30 min was 2.89% ± 0.35 %ID/g (%ID/g is percentage injected dose per gram tissue). The highest 6-OHDA dose of 100 mg/kg caused severe cardiac denervation, decreasing both NET Bmax and HED HU to 8% of their control values. Comparing values for all doses of 6-OHDA, HED retention had a strong linear correlation with NET density: HU = 0.0077Bmax −0.028, r2 = 0.95. Conclusion: HED retention is linearly dependent on NET density in rat hearts that have been chemically denervated with 6-OHDA, suggesting that HED retention is a good surrogate measure of NET density in the rat heart. This finding is discussed in relation to clinical observations of the dependence of HED retention on cardiac nerve density in human subjects using PET.
The radiotracer 11C-meta-hydroxyephedrine (HED) was developed as a clinical tool for noninvasive studies of cardiac sympathetic innervation using PET (1). A structural analog of the neurotransmitter norepinephrine, HED accumulates into presynaptic sympathetic nerve terminals through the same reuptake mechanism that recovers exocytotically released norepinephrine from the synaptic cleft. This reuptake mechanism, originally termed “uptake-1” by Iversen (2), primarily involves the transport of norepinephrine from outside the neuron into the neuronal axoplasm by the norepinephrine transporter (NET). A second transporter localized in the membranes of norepinephrine storage vesicles, the vesicular monoamine transporter (VMAT2), rapidly transports norepinephrine from the axoplasm into the vesicles for subsequent reuse by the neuron.
Previous PET studies with HED have demonstrated significant abnormalities in cardiac sympathetic nerve populations in patients with congestive heart failure (3–5), diabetic autonomic neuropathy (6–8), myocardial infarction (9), and cardiac arrhythmias (10,11). Although the ability to detect regions of cardiac sympathetic denervation with HED and PET is well documented, the precise functional relationship between semiquantitative measurements of cardiac HED retention and sympathetic nerve density needs further clarification. Without a thorough understanding of the relationship between HED retention and nerve density, it is not possible to make an accurate interpretation of these PET studies of cardiac sympathetic innervation.
In this work, a rat model of cardiac denervation was used to examine the relationship between cardiac HED retention and sympathetic nerve density, as reflected by cardiac NET density. In rats, varying degrees of cardiac sympathetic nerve denervation were produced chemically by administering different doses of the sympathetic nerve neurotoxin 6-hydroxydopamine (6-OHDA) (12). Whole-heart and regional HED retention values in vivo at 30 min after intravenous injection of the radiotracer were measured for each animal. Whole-heart NET densities, determined in vitro as the maximal binding of the NET-selective radioligand 3H-mazindol, were also measured. The results of these studies show that cardiac HED retention is linearly dependent on NET density in the rat heart. This finding is compared with existing information about the dependence of HED retention on sympathetic nerve density in the human heart.
MATERIALS AND METHODS
Radiosynthesis of 11C-meta-Hydroxyephedrine (HED)
HED was synthesized by N-methylation of (–)-metaraminol as the free base, using previously described methods (1) with a few modifications. Briefly, 11C-methyl triflate (13) in a N2 carrier gas stream was bubbled at room temperature through 0.25 mL of N,N-dimethylformamide containing 1.0 mg of (–)-metaraminol until the radioactivity trapped in the reaction vial reached a maximum (∼4 min). Because of the high reactivity of 11C-methyl triflate, the reaction to form HED is extremely rapid and no heating of the reaction vial is necessary. HED was purified using high-performance liquid chromatography (HPLC) (Phenomenex Partisil 10-μm SCX 4.6 × 250 mm column; mobile phase, 60 mmol/L NaH2PO3; flow rate, 3.0 mL/min; retention time ∼ 6 min). Total synthesis time, including HPLC purification and product formulation, was ∼35 min. Corrected radiochemical yields were ∼40%, with radiochemical purity >98% and specific activity >18.5 TBq/mmol (>500 Ci/mmol).
Cardiac Uptake of HED
All animals used in this study were cared for in accordance with the Animal Welfare Act and the National Institutes of Health's Guide for the Care and Use of Laboratory Animals (14). Experimental protocols involving animals were approved by the University Committee on Use and Care of Animals at the University of Michigan. A group of 30 male Sprague–Dawley rats, with body weights ranging from 216 to 302 g, were divided into 6 subgroups (n = 5 each). A different dose of 6-OHDA was administered to each subgroup of rats: 0 (controls), 7, 11, 15, 22, and 100 mg/kg (intraperitoneally). For each 6-OHDA dose level, the injectate was prepared by dissolving a sufficient amount of 6-OHDA into a sterile 0.9% sodium chloride solution so that the volume injected intraperitoneally into each animal was in the range of 0.4–0.8 mL. For the control subgroup, 0.5 mL of sterile 0.9% sodium chloride injectate was administered to each animal. Twenty-four hours after 6-OHDA administration, each animal was injected intravenously with 3.7–8.3 MBq (100–225 μCi) of HED. The animals were killed by decapitation while under anesthesia 30 min after HED administration and their hearts were removed and cut into 4 sections: left atrium, right atrium, left ventricle, and right ventricle. Heart sections and a whole blood sample were placed into preweighed tubes. The tubes were weighed again to determine tissue mass and placed on ice until they were counted in a γ-counter (MINAXI Auto-γ-5500; Perkin-Elmer). The γ-counter data were corrected for radioactive decay and normalized to tissue mass to determine HED tissue concentrations, expressed as percentage injected dose/g tissue (%ID/g). The entire study was performed by staggering the 6 subgroups over a 3-d period (2 subgroups per day).
Measurement of Cardiac NET Density
After counting the heart and blood samples, each animal's heart sections were placed together in aluminum foil, snap frozen in liquid nitrogen, and stored at −80°C. For the NET density assay, hearts were later processed (tissue homogenization, differential centrifugation) into a membrane preparation using previously described methods (15). Whole-heart NET density (maximum number of binding sites [Bmax], fmol/mg protein) was measured for each heart using a saturation binding assay with 3H-mazindol as the radioligand, as previously described (15).
Chemicals
6-Hydroxydopamine hydrobromide and all chemicals used to prepare buffers for the saturation binding assays were purchased from Aldrich Chemical Co. [4′-3H]Mazindol, with specific activity 777 GBq/mmol (21.0 Ci/mmol), was purchased from Perkin Elmer/New England Nuclear (product no. NET-816).
Data Analysis and Statistics
Nonlinear regression analyses were performed using GraphPad Prism software (GraphPad Software Inc.). The saturation binding assay data were fit to a 1-site binding model, B = (Bmax·L*)/(KD + L*), where B is the bound radioligand concentration and L* is the free radioligand concentration. Statistical tests for significant differences between measurements were performed using the Student 2-tailed t test for 2 samples with equal variance, with P values < 0.05 considered to be significant.
RESULTS
Mean values of cardiac NET densities (Bmax), 3H-mazindol binding affinities (KD), and HED concentrations in heart and blood at 30 min for each dose of 6-OHDA are presented in Table 1. Included are regional measurements of HED retention at 30 min in the atria and ventricles.
Averages of NET Bmax, 3H-Mazindol Binding Affinity (KD), and HED Tissue Concentrations
Cardiac NET Density
Representative saturation binding assay data for each 6-OHDA dose are given in Figure 1, along with the corresponding estimates of cardiac NET density (Bmax) and 3H-mazindol binding affinity (KD) obtained from the nonlinear regression analysis. In controls, cardiac NET densities (Bmax) averaged 388 ± 23 fmol/mg protein and 3H-mazindol binding affinity (KD) was 0.72 ± 0.07 nmol/L. NET densities decreased in a dose-dependent manner with injected 6-OHDA dose (Fig. 2A). The highest 6-OHDA dose of 100 mg/kg caused severe cardiac denervation, reducing the average NET Bmax to 31 ± 6 fmol/mg protein, a 92% reduction relative to controls (P < 5.8 × 10−10). At intermediate doses of 6-OHDA, considerable variability in measured NET densities was seen. Fitting these data to a sigmoidal dose–response model with variable slope gave an estimated median effective concentration (EC50) of 9.9 ± 2.1 mg/kg, with a Hill slope of −1.46.
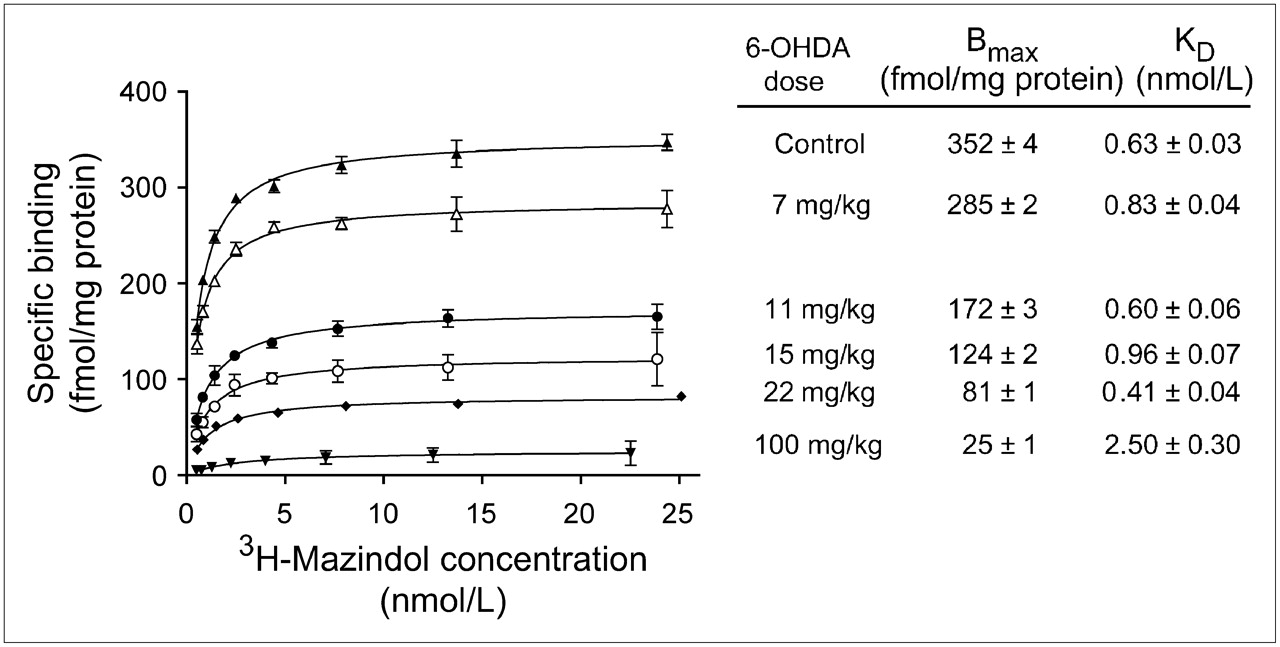
Representative saturation binding assays for different 6-OHDA doses. Specific binding data are means ± SEM of triplicate determinations at each 3H-mazindol concentration. Solid lines are nonlinear regression fits of data to a 1-site binding model. For each assay, estimated values of Bmax and KD, and the associated uncertainties in those estimates, are shown.

(A) Effect of 6-OHDA–induced denervation on cardiac NET density (Bmax). (B) Effect of 6-OHDA–induced denervation on cardiac 11C-HED uptake at 30 min. (C) Dependence of cardiac 11C-HED uptake at 30 min on NET density (Bmax).
HED Uptake
In controls, whole-heart HED uptake at 30 min averaged 2.89 ± 0.35 %ID/g. In animals treated with 6-OHDA, HED uptake also declined in a dose-dependent manner with the injected 6-OHDA dose (Fig. 2B). A fit of these data to a sigmoidal dose–response model with variable slope yielded an EC50 of 10.2 ± 2.2 mg/kg, and a Hill slope of −1.74. At the highest 6-OHDA dose of 100 mg/kg, the average whole-heart HED uptake was reduced 92% relative to controls to 0.22% ± 0.06 %ID/g (P < 1.6 × 10−7). As with the NET Bmax measurements, at intermediate doses of 6-OHDA a wide range of HED uptake values was seen. Plotting each heart's measured HED uptake (HU) value versus the corresponding cardiac NET density measurement (Fig. 2C), a strong linear correlation was found (HU = 0.0077Bmax − 0.028, r2 = 0.95).
Because the levels of cardiac denervation achieved at intermediate doses of 6-OHDA were highly variable, averages of HED uptake were also calculated for hearts that had comparable NET densities (Table 2). This resulted in averages with much lower SD. Note that the SDs of these averages are influenced by the number of hearts that happened to fall into each range of NET Bmax values.
Averages of NET Bmax, KD, and HED Concentrations for Hearts with Comparable NET Densities
DISCUSSION
The goal of this study was to determine the functional relationship between cardiac HED retention and sympathetic nerve density in a rat model of cardiac denervation. The neurotoxin 6-OHDA causes selective destruction of cardiac sympathetic nerve terminals in rats after its systemic administration through intraperitoneal or intravenous injection (12). The selectivity of 6-OHDA for sympathetic nerve terminals in the periphery is due to its efficient uptake into sympathetic neurons through the NET, since sympathetic nerve damage is prevented when NET-selective inhibitors such as desipramine are administered before 6-OHDA injection (16). Once inside sympathetic neurons, 6-OHDA undergoes autooxidation and conversion into reactive oxygen species (quinones, peroxides, superoxide radical), which form covalent bonds with structural lipids, sulfhydral groups of proteins and enzymes, amino groups of proteins, and other structural groups to cause destruction of sympathetic nerve varicosities (17). In this study, a range of 6-OHDA doses was used to cause varying levels of cardiac sympathetic denervation in rats. Cardiac HED retention was measured 24 h after 6-OHDA injection as previous studies have demonstrated that 6-OHDA–induced damage to cardiac sympathetic nerves is complete by this time (12,17). In addition, we previously showed that NET density in the rat heart is decreased in a dose-dependent manner at 24 h after intraperitoneal injections of 6-OHDA (15). HED retention was measured at 30 min after tracer administration. This time point was chosen because heart and blood levels of HED have essentially reached constant levels by 30 min in the rat (1), and because it provided maximal counting statistics for the regional tissue concentration measurements. This time is comparable to the 30- to 40-min PET image frame frequently used for calculating HED retention in clinical PET studies (18).
Whole-heart cardiac HED retention and NET density each followed sigmoidal dose–response curves with injected 6-OHDA dose, exhibiting similar EC50 values of ∼10 mg/kg. Measurements of both HED retention and NET density tended to be highly variable at intermediate doses of 6-OHDA. This variability may be due to variable absorption of 6-OHDA from the gut in different animals, leading to varying degrees of cardiac denervation (17). However, even when equivalent doses of 6-OHDA are directly injected into the brains of rats to cause partial denervation of striatal dopaminergic neurons, highly variable degrees of striatal denervation are achieved (19). Thus, the variable levels of cardiac denervation we observed at intermediate doses of 6-OHDA are consistent with previous studies with this compound. Regardless of the specific cause for the variable responses at intermediate 6-OHDA doses, the measured NET Bmax values and HED uptake values had a strong positive covariance. For a given dose of 6-OHDA, the hearts with high NET Bmax were also the ones with high HED retention, and the ones with low NET Bmax had low HED retention. This was true over the entire range of 6-OHDA doses, and a strong linear correlation was found to exist between cardiac HED retention and NET density in the rat heart. These results demonstrate that the widespread damage to sympathetic nerve varicosities caused by 6-OHDA includes the destruction of functional NET proteins. The observed linear relationship between HED retention and NET density suggests that the retention of HED in the rat heart is predominantly determined by regional NET density.
Blood values of HED were 40%–45% above control values in animals treated with 6-OHDA doses of 11, 15 and 22 mg/kg (P < 0.001). These modestly higher blood values at 30 min did not appear to have a significant effect on HED retention, consistent with previous observations that HED blood levels fall rapidly and cardiac HED uptake peaks within 5 min after HED injection (1). We did not measure blood levels of HED metabolites in this study, but since HED is primarily metabolized in the liver (20), it is unlikely that metabolism of HED would be greatly affected by cardiac denervation, given that no more than 4% of the injected dose ends up in the heart (1). We also did not measure plasma catecholamine levels in this study, but a previous report showed that administration of 6-OHDA to Sprague–Dawley rats decreases plasma norepinephrine levels by 46% while increasing plasma epinephrine only 2-fold (21). Thus, we do not believe that our HED retention measures were affected by any significant changes in HED metabolism or by competition from plasma catecholamines.
Do our results showing a linear relationship between HED retention and nerve density in rats treated with 6-OHDA translate to clinical studies of cardiac denervation in human subjects? Little information exists on the relationship between HED retention and nerve density in human heart, primarily because there is no validated noninvasive measure of regional cardiac nerve density that can be used as a gold standard. In the only study that has directly measured HED retention as a function of NET density in human heart, Ungerer et al. studied 8 patients with severe heart failure who were awaiting heart transplantation (22). Cardiac PET studies with 13N-ammonia and HED were performed <3 mo before the patients underwent heart transplantation. Myocardial perfusion was essentially normal in all patients, but regional HED deficits were seen, primarily in the lateral and inferior apical walls. After transplantation surgery, cardiac tissue samples from 9 different regions of the explanted heart were assayed for NET density. Averaging regional values across all 8 patients, HED retention was found to be linearly correlated with NET density. However, whereas NET Bmax values for the 9 regions ranged from a high of ∼283 fmol/mg protein to a low of ∼89 fmol/mg protein (69% lower than the high value), corresponding HED retention values were lower by only 20%. Thus, HED retention did not decrease proportionally with NET Bmax values in these heart failure patients and, hence, was an insensitive measure of regional NET density. These findings are not consistent with our observations of a strong positive covariance between HED retention and NET density in the rat heart but, instead, suggest a nonlinear relationship between HED retention and sympathetic nerve density in the human heart.
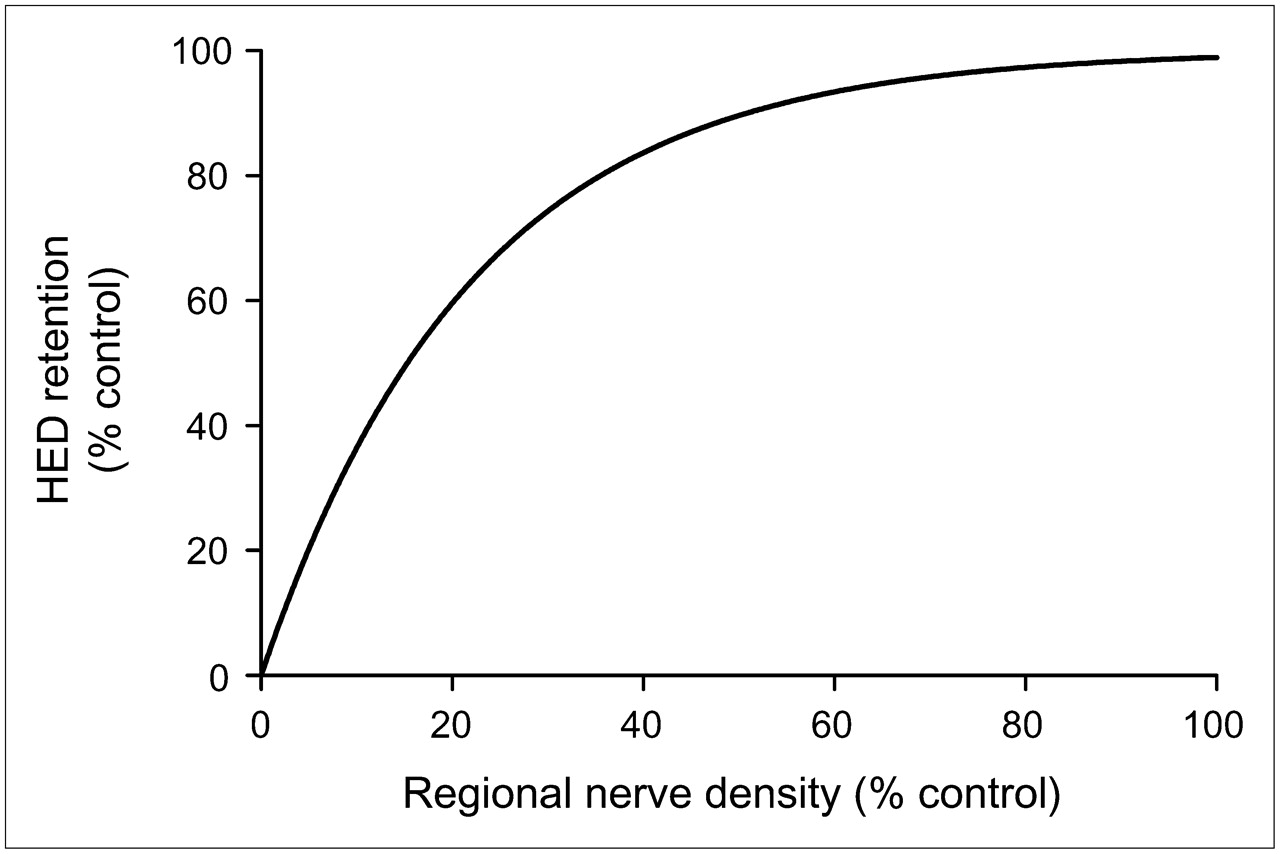
Why might HED retention be nonlinearly related to NET density in human heart? We hypothesize that the rapid neuronal uptake of HED causes regional HED retention measurements to be insensitive to low-to-moderate levels of denervation in human heart. On the basis of our interpretation of previous studies in animals and humans (18), a comprehensive compartmental model of myocardial HED kinetics is presented in Figure 3. Because the NET transport rate of HED (k3 in Fig. 3) is much faster than the rate of clearance back into plasma (k3 ≫ k2), most of the HED molecules delivered from plasma to the extracellular space are rapidly transported into the neurons. This causes the neuronal uptake of HED to be rate limited by delivery from plasma into interstitium (K1), rather than by NET transport (k3). Since delivery of HED into interstitium from plasma is determined by the product of its unidirectional extraction fraction (E) and blood flow (F)—that is, K1 = E·F—the rapid NET transport of HED causes it to be a delivery-limited or flow-limited tracer. Supporting this view, HED retention was previously shown to be flow dependent in the dog heart (23). With neuronal uptake of HED rate limited by delivery, regions with low or moderate denervation would have sufficiently large NET populations for the surviving neurons to accumulate most of the HED molecules delivered from plasma. In such regions there would be little apparent change in the myocardial kinetics of HED. Thus, for a flow-limited sympathetic nerve tracer, tracer retention would be insensitive to nerve losses early in the course of denervation and would decline only after nerve losses became severe. In this case, there would be a nonlinear relationship between HED retention and nerve density, as shown in Figure 4. This type of nonlinear relationship is consistent with the findings of Ungerer et al. in heart failure patients discussed earlier (22).
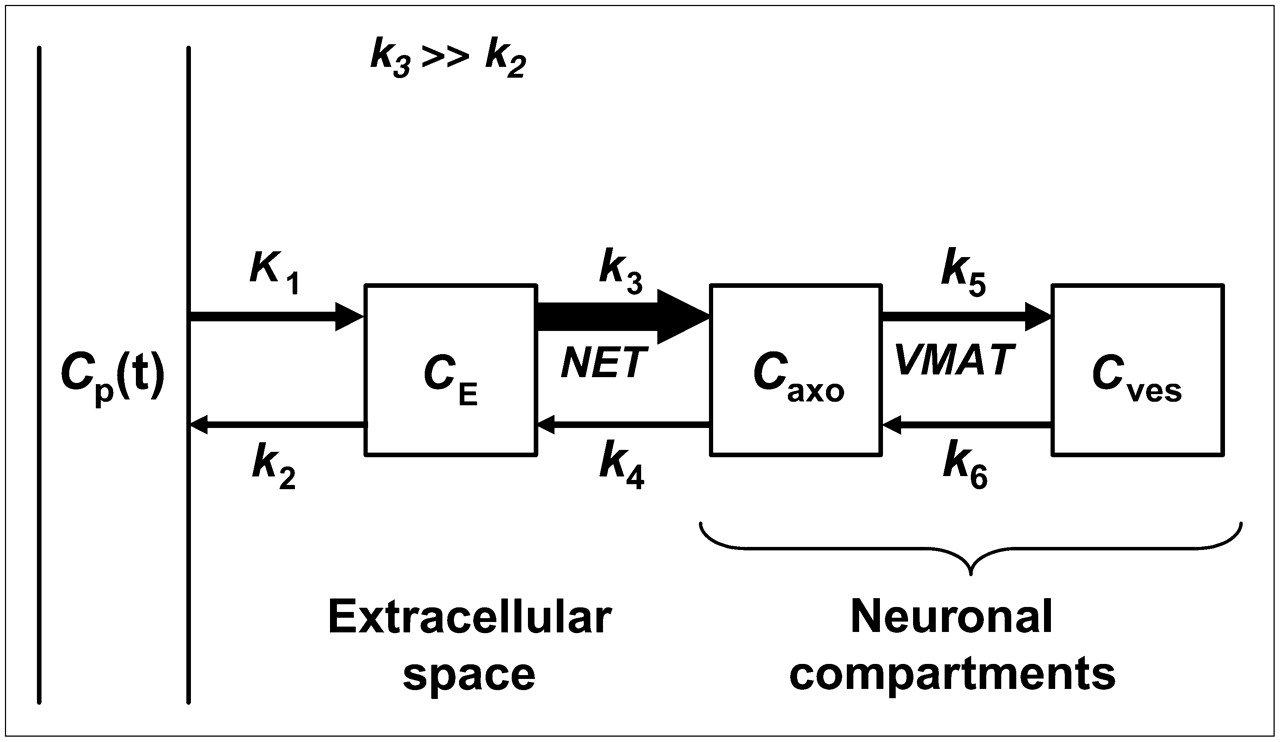
Proposed comprehensive compartmental model of cardiac HED kinetics. Arrow thicknesses indicate relative magnitudes of the rate constants. Cp = concentration in plasma; CE = concentration in extracellular space; Caxo = concentration in neuronal axoplasm; Cves = concentration in vesicles.

Expected nonlinear relationship between HED retention and regional sympathetic nerve density in human heart, based on the comprehensive compartmental model shown in Figure 3.
Why would HED have a strong linear correlation with nerve density in the rat heart but a nonlinear relationship with nerve density in the human heart? We do not currently have sufficient data to definitively address this question, but one possible explanation is that NET density in the human heart is much higher than that in rat heart. Using comparable binding assay methodology for human and rat left ventricle samples, Böhm et al. measured NET Bmax values in human heart to be ∼15 times higher than that in rat heart (1,102 vs. 71 fmol/mg protein) (24,25). The transport kinetics of NET substrates are well described by Michaelis–Menten kinetics; each substrate possesses a unique combination of the transport parameters Vmax (the maximum velocity of transport) and Km (the half-saturation concentration). For a given NET substrate concentration [S], the initial velocity of transport by NET is given by Vmax[S]/(Km + [S]). In imaging studies, only tracer-level concentrations of substrate are used (i.e., [S] ≪ Km), so the initial velocity of transport simplifies to (Vmax/Km)[S]. Thus, the neuronal uptake rate of a radiolabeled NET substrate is characterized by the ratio of its transport constants Vmax/Km. Since Vmax is directly proportional to transporter density (i.e., NET Bmax), a higher NET density in human heart relative to rat heart would be expected to lead to a faster neuronal uptake rate of HED in human heart. This faster NET uptake rate would cause HED to be flow-limited in human heart, leading to a nonlinear relationship between HED retention and NET density.
If further studies confirm a consistent nonlinear relationship between HED retention and NET density in human heart, this would have important clinical implications. It would mean that when detectable HED retention deficits are observed, they in fact reflect substantial regional nerve losses. Because detection of more modest levels of denervation early in the progression of a disease could be crucial in providing the most effective therapies to halt or reverse myocardial denervation, this would represent a significant limitation in the clinical utility of HED. It would also mean that HED could not provide sensitive measures of changes in nerve density in response to novel therapies designed to slow or reverse myocardial denervation. These limitations could not be overcome by using more complex methods of analyzing the kinetics of HED, as no matter how one approaches the kinetic modeling of a flow-limited sympathetic nerve tracer, any quantitative measures obtained will be insensitive to nerve losses until they become severe. The only way to overcome these limitations would be to develop a new tracer possessing superior kinetic properties for measuring regional sympathetic nerve density. Specifically, this new tracer would need to have a much slower NET transport rate than HED, so that its neuronal uptake rate is limited by NET transport rather than by tracer delivery. Referring again to Figure 3, ideally this tracer would have a NET transport rate k3 that is ≤ k2. In this case, the measured cardiac retention of the tracer would be much more sensitive to low-to-moderate levels of denervation, and tracer retention would follow a more linear relationship with nerve density. In light of these considerations, we believe that development of novel sympathetic nerve radiotracers with more optimal kinetic properties for quantifying sympathetic nerve density is warranted.
CONCLUSION
Cardiac retention of HED was found to have a strong linear correlation with NET density in a rat model of cardiac denervation. These results improve our understanding of the sensitivity of HED retention to denervation in the rat heart, which will aid in the interpretation of studies that use HED and rat models to investigate the effects of diseases on cardiac sympathetic innervation. Further work is needed to better define the functional relationship between HED retention and regional cardiac sympathetic nerve density in human subjects.
Acknowledgments
We thank the staff of the University of Michigan Cyclotron/PET Facility, especially Louis Tluczek, for their assistance in preparing HED. We also thank Robert Koeppe for his critique of the manuscript. This work was supported by grants R01 HL079540 and R29 HL59471 from the National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD.
Footnotes
-
COPYRIGHT © 2006 by the Society of Nuclear Medicine, Inc.
References
- Received for publication March 17, 2006.
- Accepted for publication May 10, 2006.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Sympathetic Innervation of Cold-Activated Brown and White Fat in Lean Young Adults
- Mechanistic Insights into Sympathetic Neuronal Regeneration: Multitracer Molecular Imaging of Catecholamine Handling After Cardiac Transplantation
- Multiparametric Molecular Imaging Provides Mechanistic Insights into Sympathetic Innervation Impairment in the Viable Infarct Border Zone
- Presence of Specific 11C-meta-Hydroxyephedrine Retention in Heart, Lung, Pancreas, and Brown Adipose Tissue