


Abstract
This study evaluated the potential use of dynamic PET to monitor transient metabolic processes and to investigate the mechanisms of action of new photosensitizing drugs in the photodynamic therapy (PDT) of cancer. Methods: Rats bearing 2 mammary tumors received different phthalocyanine-based photosensitizers. The following day, the animals were positioned in a Sherbrooke small-animal PET scanner and continuously infused with 18F-FDG while dynamic images were acquired for 2 h. During that period, one of the 2 tumors was exposed for 30 min to red light delivered by a small diode laser to activate PDT. Results: 18F-FDG time–activity curves during PDT showed distinct transient patterns characterized by a drop and subsequent recovery of tumor 18F-FDG uptake rates. Variations in these rates and response delay parameters revealed tumoral and systemic metabolic processes that correlated with differences in mechanism of action between drugs, that is, direct tumor cell kill or initial vascular shutdown. Conclusion: Real-time follow-up of tumor response to PDT as monitored by dynamic 18F-FDG PET has been shown to correlate with the mechanisms of action of photosensitizing drugs in vivo. This new imaging paradigm can be exploited to monitor a variety of transient cellular and molecular processes as they occur in vivo, enabling the mechanisms of action of therapeutic interventions to be scrutinized and their efficacy predicted in real time.
Photodynamic therapy (PDT) is a promising treatment for light-accessible tumors (1). This type of therapy combines tumor-localizing photosensitizers, visible light, and molecular oxygen to induce oxidative damage to the tumor tissue (2). Most clinical applications of PDT use porfimer sodium (Photofrin; Axcan Pharma PDT Inc.) (3), a mixture of hematoporphyrin derivatives, but second-generation photosensitizers such as the metallophthalocyanines (Pc) with better photochemical properties than porfimer sodium have been developed (4). Phthalocyanines form stable chelates with metal cations such as Zn2+ and Al3+, and the addition of sulfonate groups at the periphery of the macro cycle gives water-soluble photosensitizers with well-documented potential for use in clinical PDT (5). The results of many studies of PDT in rodents have provided evidence for 2 types of response mechanisms that lead to tumor regression (6). PDT may cause direct tumor cell kill or activate endothelial cells to release vasoactive compounds that provoke vascular stasis followed by indirect damage to malignant cells. In addition to cell or vascular damage, release of vasoactive components may produce an inflammatory response into the tumor and surrounding tissues. The relative contribution of the 2 main pathways to overall tumor response depends on the distribution of the photosensitizer among cellular or vascular compartments, which in turn depends on the chemical nature of the photosensitizing drug (7). More hydrophobic or amphophilic photosensitizers such as disulfonated ZnPcS2 are preferentially transported by lipoproteins, which are taken up directly by tumor cells, whereas hydrophilic photosensitizers such as tetrasulfonated AlPcS4 are transported largely by albumin and deposited mainly in the vascular stroma of tumors (8). It follows that ZnPcS2-PDT induces largely direct cell kill, whereas with AlPcS4-PDT, the tumor vascular system is initially affected and indirect tumor cell death occurs afterward (9,10).
Small-animal PET is an excellent in vivo method to investigate various processes in molecular biology, oncology, and neuroscience research (11–14). In oncologic PET studies, the most widely used radiopharmaceutical for the evaluation of tumor glucose metabolic rates is 18F-FDG (15,16). 18F-FDG PET appears also to be a promising method to study the effect of PDT in vivo, either to assess the efficacy of a photosensitizing drug or to determine its mechanism of action (17,18). Our previous study showed that the tissue uptake pattern of 18F-FDG and the reduced metabolic activity of the treated tumor measured by PET at different times after PDT correlated with the mechanism of action of PDT through indirect vascular stasis or direct cell kill (17). We observed, in particular, that the effect of the PDT drug on the vascular system caused an early drop of 18F-FDG uptake in the treated tumor, within less than 30 min after the end of the PDT light treatment. In contrast, the direct effect of the PDT drug on tumor cells appeared to be delayed by about 2 h after treatment.
Whereas these studies provided clear evidence of dissimilar modes of action for the 2 different PDT drugs, the simple measurement of 18F-FDG tumor uptake at 2 time points gave little insight into the actual mechanisms of action leading to inactivation of the tumor cells. Further investigations of the tumor metabolism at specific intervals after illumination by the Patlak method confirmed these findings (19) but were of little help in unraveling the biologic processes that occur during and immediately after the light treatment and result in tumor cell obliteration. The reason they were of little help is due to 2 major problems with the conventional dynamic PET approach after a bolus injection of the radiotracer. First, the long uptake period of 18F-FDG by tumors (>30 min) does not allow an instantaneous measurement of the metabolic state of tumor tissues but, rather, represents some average value over a specified interval. This is fine for a tumor (or any other tissue) in a steady state but is confounding for tissues undergoing rapid transient metabolic processes. The second related problem is that the effect of PDT treatment continues to evolve during the 18F-FDG uptake period, hence preventing any instantaneous transient metabolic processes from being clearly differentiated.
This study explored the potential of using real-time dynamic 18F-FDG PET with constant radiotracer infusion as a means to evaluate the role of the photosensitizer in the PDT tumor response mechanism. The photosensitizers selected for this study are known to induce tumor regression via different pathways, that is, via direct tumor cell kill and via indirect tumor cell kill for ZnPcS2 photosensitizers and AlPcS4 photosensitizers, respectively.
MATERIALS AND METHODS
Photosensitizers
Adjacently disulfonated ZnPcS2 and tetrasulfonated AlPcS4 (Fig. 1) were synthesized by a condensation method previously detailed (20). A few milligrams of Pc were dissolved in phosphate-buffered saline (PBS), 1 mmol/L, and sonicated for a few minutes. The solutions were filtered on Millex-GV, 0.22 μm (Millipore), under sterile conditions. The concentration of the dye solutions was determined by ultraviolet and visible absorption spectroscopy after dilution in dimethyl formamide (ε = 2.5 × 10−5 (mol/L)−1cm−1 at λ = 675 nm). The working solutions were prepared by diluting the stock solutions in PBS such as to give final drug doses of 1 and 7.2 μmol/kg for ZnPcS2 and AlPcS4, respectively, in a total volume of 1 mL/100 g of body weight.
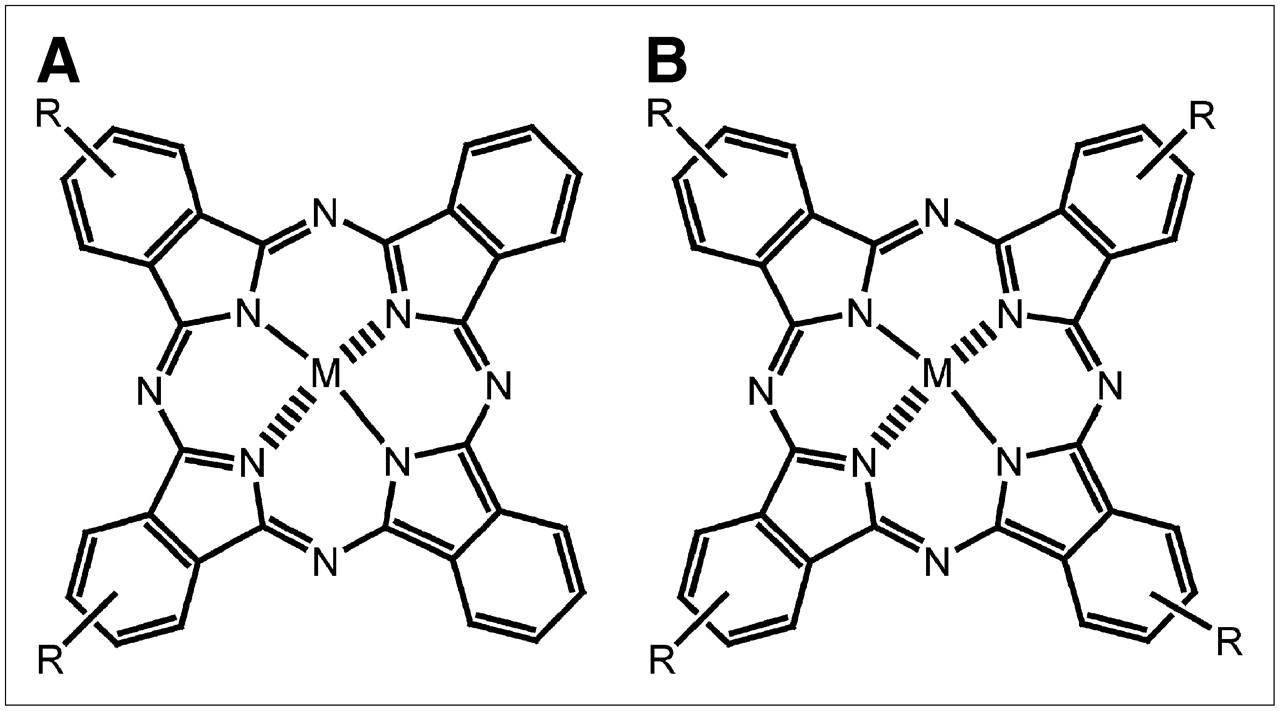
Chemical structure of ZnPcS2 (A) and AlPcS4 (B). M in A and B = zinc and aluminum, respectively; R = SO3.
Animal Model
All experiments were performed on Fischer 344/CRBL female rats (150−160 g) (Charles River Breeding Laboratories) bearing two 13762 mammary adenocarcinoma tumors. The experiments followed a protocol approved by the Canadian Council on Animal Care and the in-house ethics committee. Animals were allowed free access to water and food throughout the experiments, except for the night preceding the 18F-FDG PET study, when they were kept fasting.
Tumor Implantation
Mammary adenocarcinoma tumor cells (2.25 × 106 cells in 0.15 mL of PBS) were intradermally implanted in both axillary areas of each rat. The 2 tumors were allowed to grow 7 d before the PDT PET studies (tumor size: length [l] = 14.4−15.1 mm; width [w] = 9.8−11.9 mm; thickness [h] = 7.6−8.2 mm). Tumor volume was measured externally with an electronic caliper according to a hemiellipsoid model (1/2 [4π/3] × [l/2] × [w/2] × h). No tumor necrosis was evident at the time of treatment.
PDT
The day before the PDT PET study, the rats were given 1 μmol of ZnPcS2 or 7.2 μmol of AlPcS4 per kilogram of body weight through the tail vein (in 1 mL of PBS/100 g of body weight), representing the minimal dye dose required for total tumor regression after PDT. The PDT light treatment was applied 24 h after dye administration during the 18F-FDG PET study. One tumor was illuminated with a 670-nm light beam delivered via a fiber optic by a diode laser (model BWF-670-300; B&W Tek, Inc.), and the other tumor was masked and served as a control. The light beam was spread uniformly over the whole tumor area and maintained for 2,000 s at a fluence rate of 200 mW/cm2, to give a total fluence of 400 J/cm2.
PET Studies
PET was performed with the Sherbrooke small-animal PET scanner (21). The scanner is made of 2 avalanche photodiode detector rings and produces 3 image planes (2 direct, 1 cross) over a 1-cm axial field of view. The scanner has a flexible system of acquiring list-mode data that allows elaborate dynamic PET image series to be extracted as desired. The rats were anesthetized (2.5% isoflurane in medical O2), and a butterfly cannula (Butterfly-25 Short; Venisystems) was placed in the tail vein. The position of the scanner bed was adjusted in order to place the tumors at the center of the axial field of view with the help of a laser pointer. Thirty minutes before the onset of illumination, a continuous infusion of 18F-FDG (∼250 MBq in 0.96 mL of PBS at 0.008 mL/min) was started while a 2-h dynamic PET image acquisition was launched. One tumor was illuminated for 2,000 s, and imaging was then continued for about another hour to complete the total 2-h scan sequence. The vital signs of the animals were monitored and recorded throughout the 2 h to ensure a stable physiologic status at all times. PET images consisting of 120 frames of 60 s each were sorted out from the list-mode data for each of the 3 planes and reconstructed by a maximum-likelihood expectation maximization algorithm implementing detector response (22). Four rats were scanned for each photosensitizer. Controls included scans of 2 rats that received photosensitizer but no light and scans of 2 rats that received light but no photosensitizer.
Image Analysis
Regions of interest were traced over the control and treated tumors on the last frame of the dynamic image series and applied to all preceding frames. Time–activity curves corrected for radionuclide decay were generated, and a mathematic model was used to fit 3 distinct parts of the curves corresponding to radiotracer uptake before, during, and after the illumination period. The following equations were used to fit the curves from the treated and untreated tumors: where t is the time and m1, m2, and m3 are the slope values representing tumor 18F-FDG uptake rates before, during, and after PDT illumination, respectively. The bi variables are fitting parameters that have no special meaning. Two additional parameters were introduced to describe the duration of the transient states that resulted from the intervention and were observable from the tumor 18F-FDG uptake curves: response delay Δ1, which is the time lag after the start of illumination, Ti, until the 18F-FDG uptake rate starts to drop, and recovery delay Δ2, which is the time lag after the end of illumination, Tf, until the 18F-FDG uptake rate recovers. These 2 delay parameters were extracted from the intersections of the equations fitted to the 3 steady-state parts of the tumor 18F-FDG uptake curves.
where t is the time and m1, m2, and m3 are the slope values representing tumor 18F-FDG uptake rates before, during, and after PDT illumination, respectively. The bi variables are fitting parameters that have no special meaning. Two additional parameters were introduced to describe the duration of the transient states that resulted from the intervention and were observable from the tumor 18F-FDG uptake curves: response delay Δ1, which is the time lag after the start of illumination, Ti, until the 18F-FDG uptake rate starts to drop, and recovery delay Δ2, which is the time lag after the end of illumination, Tf, until the 18F-FDG uptake rate recovers. These 2 delay parameters were extracted from the intersections of the equations fitted to the 3 steady-state parts of the tumor 18F-FDG uptake curves.
Statistical Analysis
The values reported represent the average ± SD over the number of subjects in each group. The significance of differences between mean values was determined by the Student t test.
RESULTS
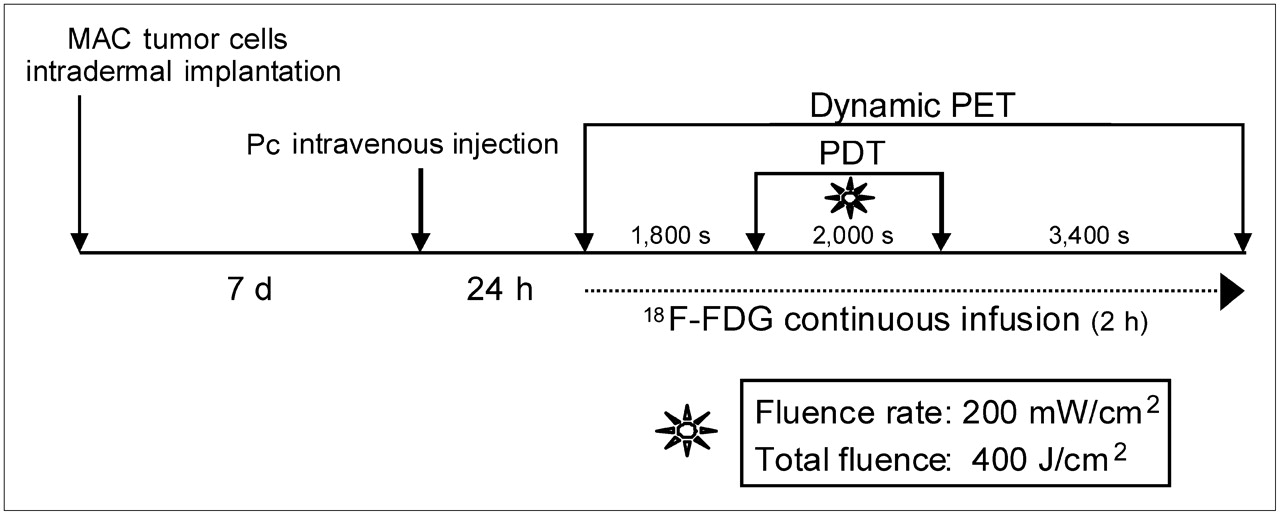
The experimental protocol used for this investigation is illustrated in Figure 2. Changes in tumor 18F-FDG uptake were visible on the PET scans (60 s each) obtained during the 2-h real-time 18F-FDG PET sequence (Fig. 3). The accumulation of 18F-FDG in treated tumor dropped significantly during the illumination period, followed by partial recovery on termination of the light treatment.
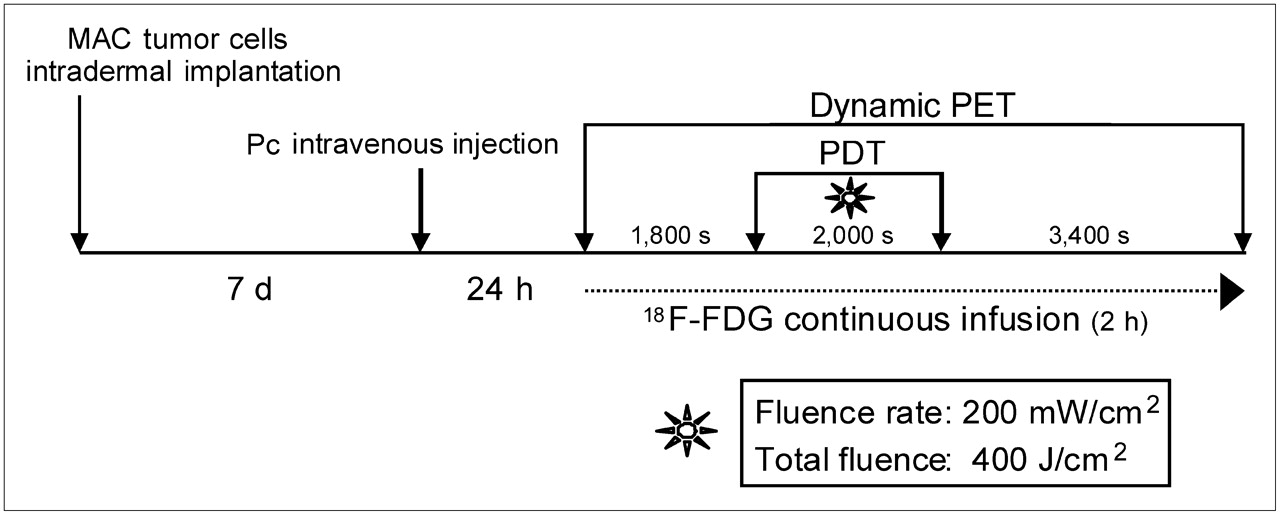
Schematic representation of steps of experimental PDT PET protocol.

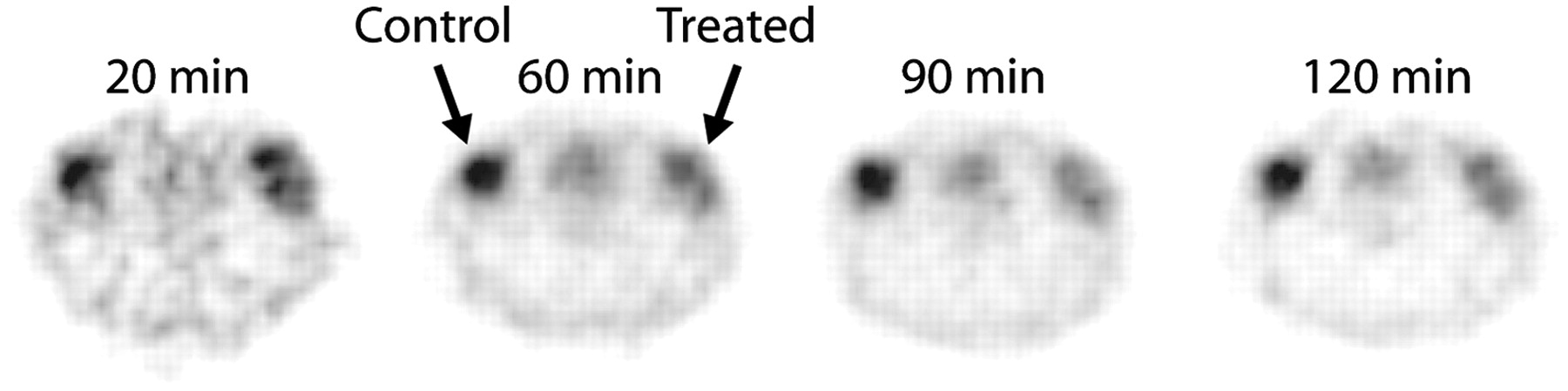
Selected 18F-FDG PET image frames (1-min duration) taken at different times during ZnPcS2-PDT treatment. Although 18F-FDG uptake is comparable in the 2 tumors before PDT treatment (20 min), uptake is significantly reduced at end of light treatment (60 min) in treated tumor relative to control. Partial recovery of 18F-FDG uptake is apparent after treatment (90 min) and even more so at end of imaging sequence (120 min).
Differences in 18F-FDG uptake between the 2 PDT protocols were more clearly visible from the 18F-FDG time–activity curves taken over the untreated and light-treated tumors (Figs. 4A and 4B). Both ZnPcS2-PDT and AlPcS4-PDT induced a drop in the 18F-FDG uptake rate followed by a partial recovery some time after the end of the illumination period, but with different response patterns. Both control scans, that is, administered photosensitizer without light treatment or light treatment without photosensitizer, showed a steady increase in 18F-FDG uptake by both tumors (Figs. 4C and 4D). The differences in baseline 18F-FDG uptake before PDT illumination resulted solely from the different tumor states at the time of the procedure.

Typical 18F-FDG tumor uptake curves during real-time 18F-FDG PET study. Two tumors were implanted in each animal, but only one tumor received light treatment. 18F-FDG uptake curves were generated for treated and untreated tumors during ZnPcS2-PDT (A), during AlPcS4-PDT (B), during control treatment with ZnPcS2 without light (C), and during control treatment without photosensitizer but with light (D).
Analysis of the 18F-FDG time–activity curves, as depicted for ZnPcS2-PDT in Figure 5, showed that the 3 distinctly different steady phases—m1, m2, and m3—and 2 transient regions characterized by response delays—Δ1 and Δ2—properly describe the 18F-FDG uptake rate. Combined, these parameters enabled clear discrimination between direct and indirect mechanisms of tumor cell kill but also provided some insight into the mechanisms of action of different drugs early during the photodynamic process.
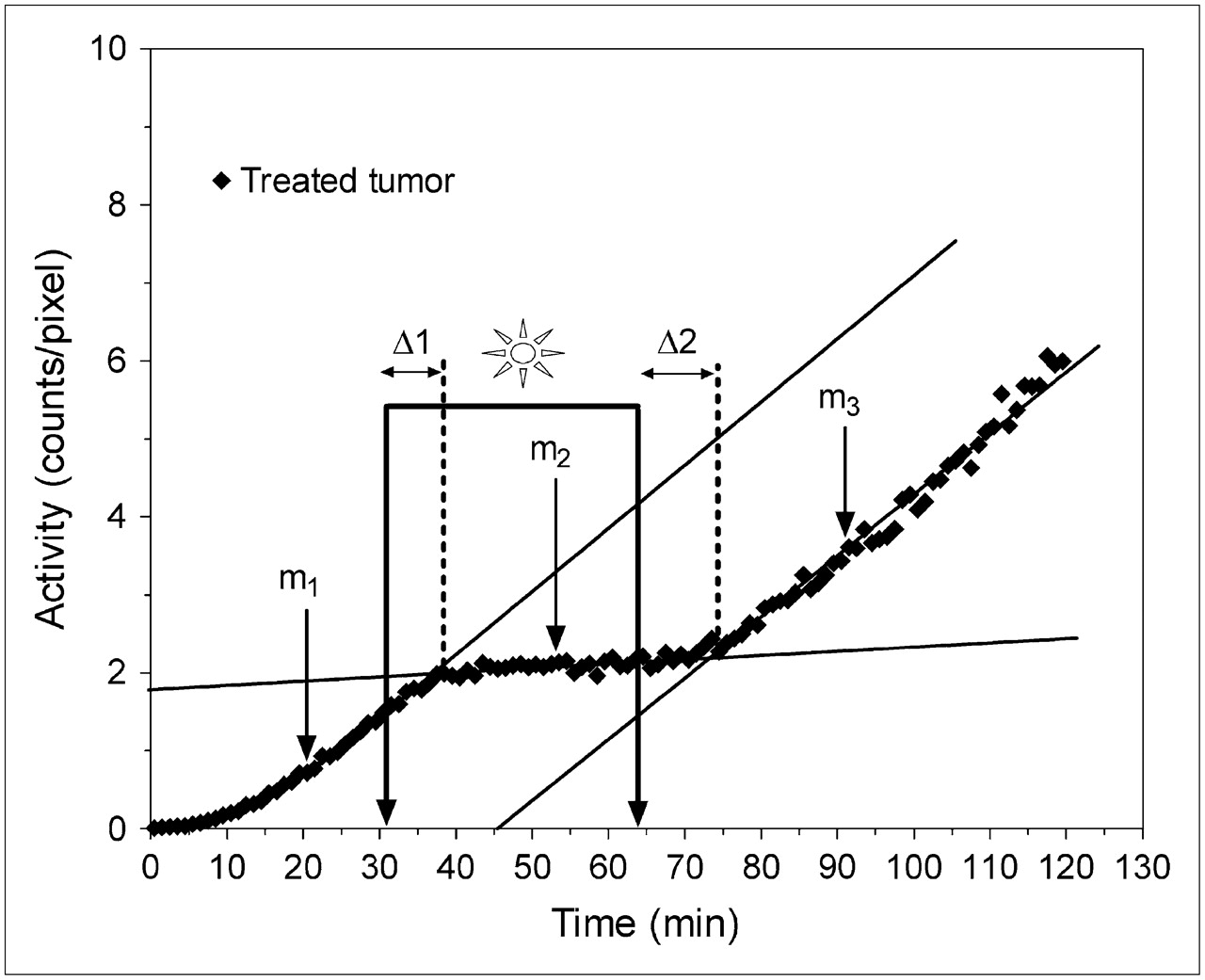
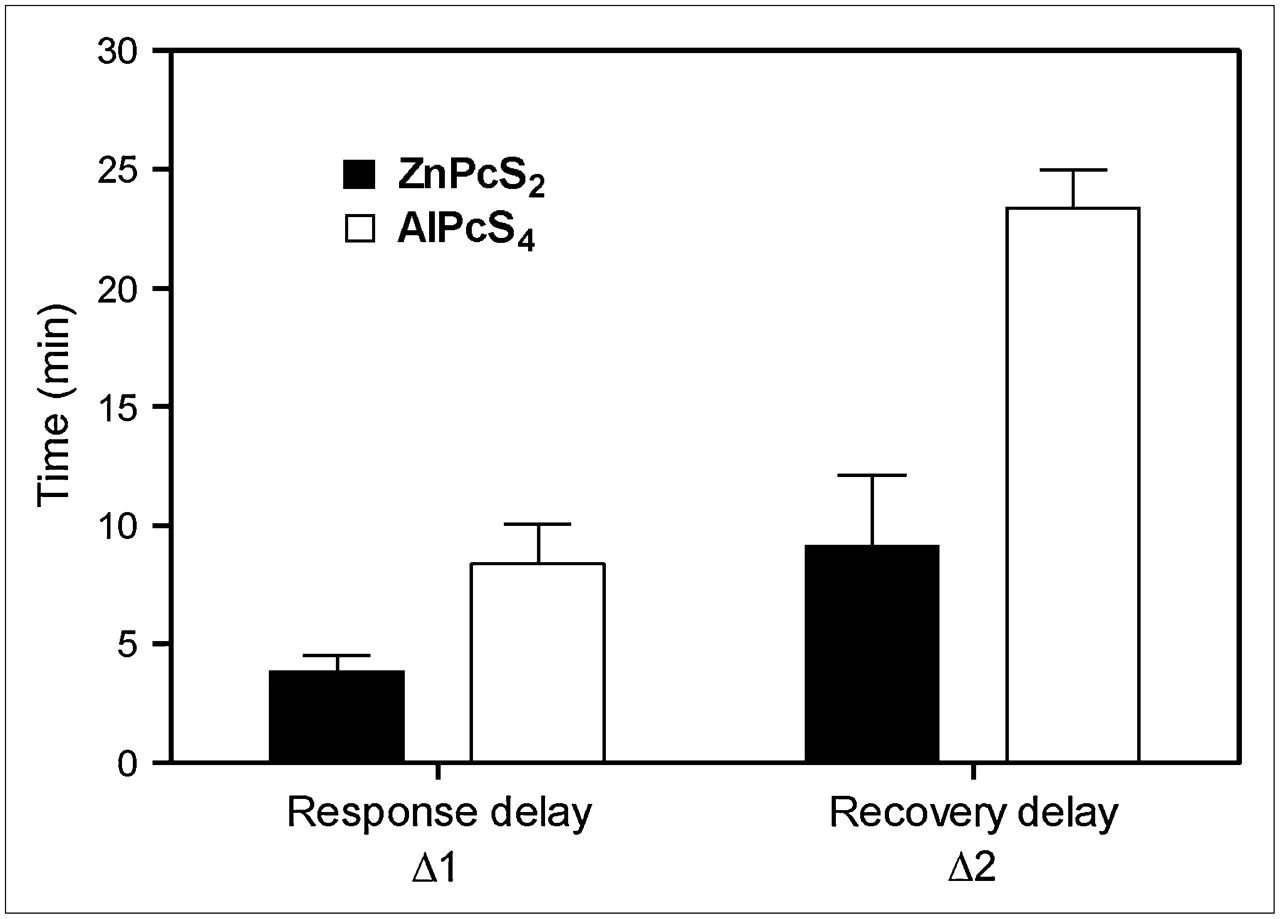
18F-FDG tumor uptake curve during ZnPcS2-PDT treatment depicting the 3 distinct 18F-FDG uptake phases and 5 parameters used to describe uptake slopes, response delay time (Δ1), and recovery delay time (Δ2).
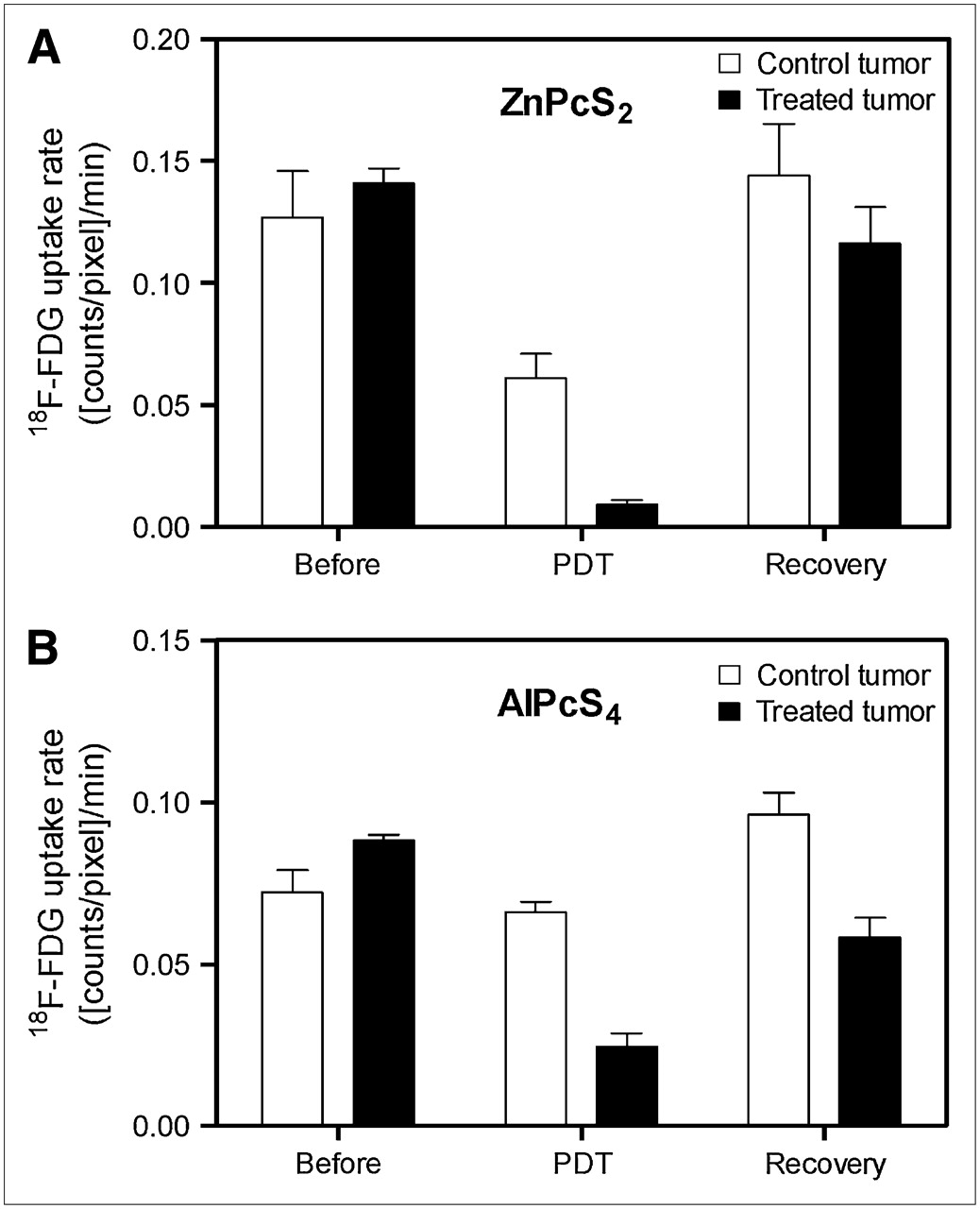
The effect of the photosensitizer on PDT tumor response is summarized by the average 18F-FDG uptake rates (Fig. 6) and response-recovery delays (Fig. 7) of 4 independent experiments. In the case of ZnPcS2-PDT, which induces substantially direct tumor cell kill, the rate dropped dramatically (>90%) within 4 min after the start of the light treatment while recovering to about 80% of the initial rate within 10 min after the end of the treatment, as is illustrated in Figure 6A for 18F-FDG uptake rates and Figure 7 for response-recovery delays. The control tumor also showed a 50% drop in the 18F-FDG uptake rate during PDT, followed by complete recovery after the treatment, suggesting a systemic response during the illumination phase (Fig. 6A). In the case of AlPcS4-PDT, which acts largely via vascular stasis, this systemic component was small (<10% drop in 18F-FDG uptake rate), although the 18F-FDG uptake rate by the control tumor showed a slight but significant (∼30%) increase after completion of the light treatment (Fig. 6B). The 18F-FDG uptake rate in the illuminated tumor with AlPcS4-PDT dropped by 70% (Fig. 6B) after a relatively long response lag of nearly 10 min (Fig. 7). Even more significant was the long lag of almost 25 min after AlPcS4-PDT (Fig. 7) before the 18F-FDG uptake rate recovered to about 60% of the initial rate (Fig. 6B).

Histograms representing mean (±SD) tumor 18F-FDG uptake rates (counts/pixel/min) before, during, and after illumination. Data were obtained from studies with ZnPcS2-PDT (n = 4) (A) and with AlPcS4-PDT (n = 4) (B).
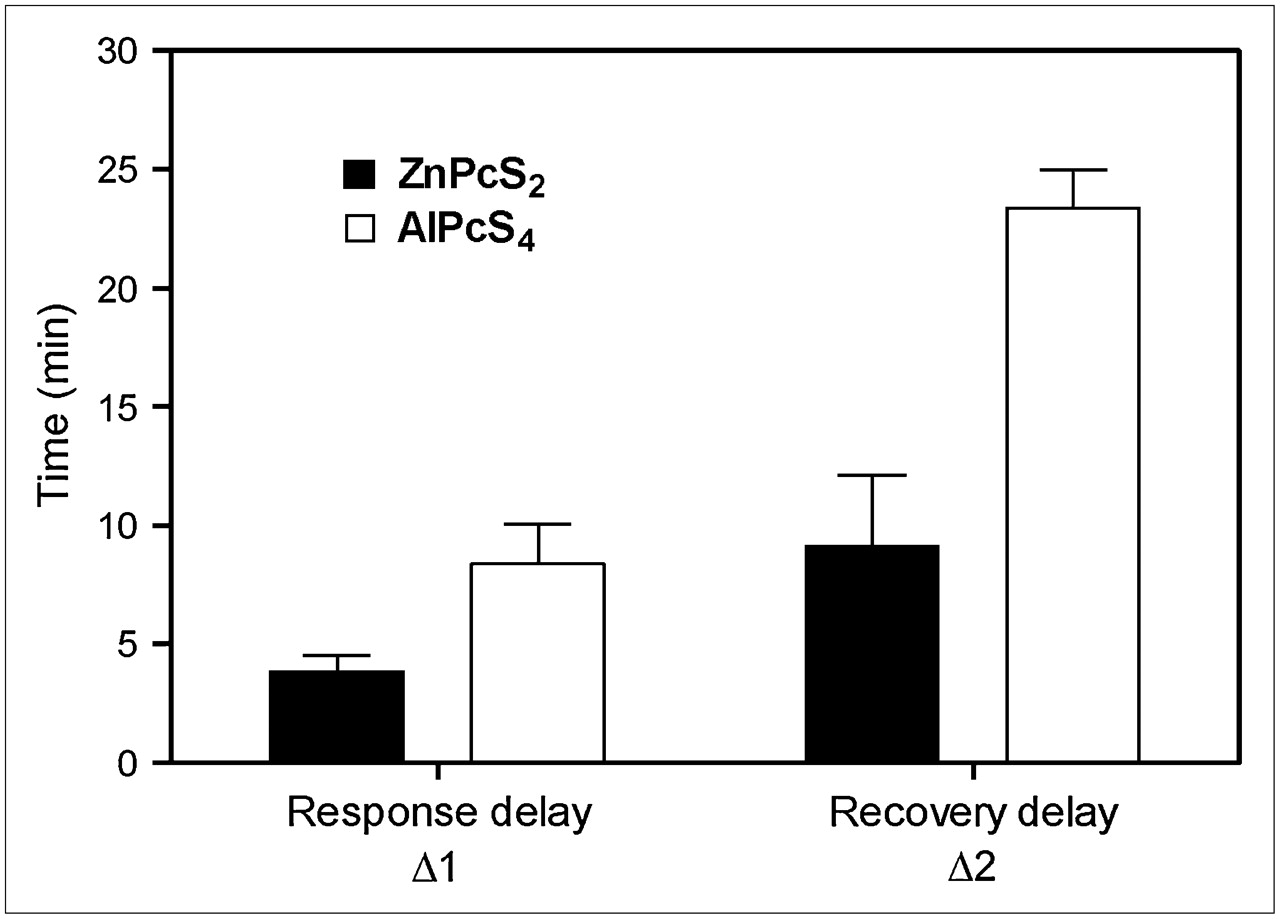
Histogram representing mean (±SD) response delay (Δ1) and recovery delay (Δ2), in minutes. Data were obtained from studies with ZnPcS2-PDT (n = 4) and with AlPcS4-PDT (n = 4).
DISCUSSION
Mechanisms of Action
PDT requires 3 components: a photosensitizer, molecular oxygen, and red light. Combined, they generate singlet oxygen, which is believed to be the principal cytotoxic entity, resulting in localized oxidative stress (23). Because of the high reactivity of singlet oxygen, this species exerts its detrimental action at the site of photosensitizer retention. Thus, the mechanism of action by which PDT induces tumor cell death depends largely on the localization of the photosensitizer during exposure to light. Chemical modifications that change the solubility of a photosensitizer without affecting its photochemical properties can significantly affect the outcome of PDT by modulating pharmacokinetics and tissue distribution. The photosensitizers selected for this study are known to target different tumor components. The amphophilic, disulfonated ZnPcS2 has good cell membrane–penetrating properties and targets subcellular membrane structures. Furthermore, binding to lipoproteins favors retention by tumor tissue through interaction with low-density-lipoprotein receptors that are overexpressed at the membrane of most tumor cells (24–26). In contrast, the highly water-soluble, tetra-substituted AlPcS4 is transported by serum albumin, resulting in accumulation within the interstitial space and vascular stroma of the tumor tissue (27). Thus, whereas ZnPcS2 readily localizes in tumor cells after intravenous injection, AlPcS4 remains in the tumor vascular compartment. The photosensitizer distribution pattern has obvious implications for the mechanisms by which PDT will induce tumor regression. PDT with ZnPcS2 impairs mitochondrial and other intracellular membrane systems, resulting in direct tumor cell kill, whereas PDT with AlPcS4 damages tumor blood vessels, interrupting the supply of oxygen and nutrients, resulting in indirect tumor cell death (28,29). In addition, varying the interval between drug administration and light exposure will affect the localization of a selected photosensitizer and, consequently, the way it will act (30). For example, PDT with the amphophilic AlPcS2 resulted in severe vascular damage to a LOX tumor model when light was applied shortly after drug injection, whereas direct tumor cell damage was observed when the illumination was performed 48 h after drug administration (31). The PDT protocol and photosensitizers used for this 18F-FDG PET study were selected such that ZnPcS2-PDT induced mainly direct tumor cell kill whereas AlPcS4-PDT provoked vascular damage.
In our experiments, changes in the 18F-FDG uptake profiles of treated tumors were more readily explained by the underlying mechanisms of action. In the case of ZnPcS2-PDT, direct PDT-induced damage to tumor cell membranes might result in a loss of glucose transporter or hexokinase activity, either of which would rapidly reduce the rate of 18F-FDG uptake by tumors. The 18F-FDG uptake rate rapidly recovered to more than 80% of the initial rate, suggesting that most of the damage was reversible (Fig. 6A). However, other factors might have contributed to this resurgence. Apoptosis is well known to be a major response to the intracellular damage inflicted by PDT (32). The fact that the apoptotic pathway also requires energy (33) could explain, at least in part, the increase in 18F-FDG uptake by the treated tumor. In addition, infiltration of inflammatory cells and the presence of residual viable tumor cells might also have contributed to maintaining the 18F-FDG uptake rate (34).
In the case of AlPcS4-PDT, damage to the vascular system was reflected in the delayed drop in the 18F-FDG uptake rate and the long recovery period of more than 20 min after the illumination ended. The recovery delay was 3 times longer than that observed in the case of the direct cell kill mechanism (Fig. 7). Also, the recovered 18F-FDG uptake rate remained substantially lower than the initial rate (Fig. 6B), suggesting that irreversible vascular damage limited blood flow and 18F-FDG transport to the tumor tissue. Again, other factors, including inflammatory cell infiltration and reversible damage, might also have contributed to the increase in the 18F-FDG uptake rate.
Other investigations using alternate, more specific radiotracers that are not subjected to secondary confounding effects such as inflammation or apoptosis would have to be considered to unravel the origin of the resurgence in 18F-FDG tumor uptake after PDT. We are currently investigating the feasibility of using the perfusion tracers 13N-ammonia and 64Cu-labeled pyruvaldehyde bis(N4-methylthiosemicarbazone) to measure tumor blood flow during and after PDT. Promising new tracers such as 18F-fluorothymidine (35) or 18F-fluoroethyltyrosine (36) could also be used with the same dynamic PET protocol as is used for 18F-FDG to measure real-time variations in cell proliferation or protein synthesis during and after PDT. The observation period could likely be extended with little modification to the protocol to determine the duration and extent of transient metabolic processes after PDT.
Systemic Response
Even when shielded from light, the control tumor in the ZnPcS2-PDT protocol showed reduced 18F-FDG uptake during the illumination phase, suggesting a systemic response originating from the treated tumor (Fig. 6A). In the case of the AlPcS4-PDT protocol, 18F-FDG uptake by the control tumor was only slightly affected during the illumination phase (Fig. 6B). However, its uptake rate increased significantly (P < 0.05) after PDT, suggesting augmented metabolic activity. Hence, the control tumors in both cases showed variations in 18F-FDG uptake resulting from the illumination phase, suggesting a systemic response originating from the treated tumors. Any uncorrelated effect of the photosensitizer or light treatment alone can be discarded, as confirmed by the control scans without light treatment and without photosensitizer (Figs. 4C and 4D). The systemic response likely results from the release of signaling factors at the site of the treated tumor. Damage to the vascular endothelium or platelets is well known to lead to production of various vasoactive compounds, including eicosanoids such as thromboxane, cytokines, clotting factors, and histamine. This in turn leads to increased vascular permeability to macromolecules, constriction of vessels, and eventually blood-flow stasis (37,38). Thus, although these vasoactive factors are released at the site of illumination, they may enter the blood circulation to act on remote blood vessels, including those of the control tumor, causing vasoconstriction and the observed drop in 18F-FDG uptake by the distant tumor.
Although this series of events has been shown to occur with photosensitizers localizing in the vascular stroma of tumors, that is, those acting via an indirect mechanism, photosensitizers acting directly on tumor cells may have a similar effect. For instance, PDT of hamster ovary cells in vitro resulted in loss of cell integrity and the release of inflammatory and immune mediators, including eicosanoids and histamine (39). Therefore, both AlPcS4-PDT– and ZnPcS2-PDT–induced release of vasoactive molecules may explain, at least in part, the decrease in 18F-FDG uptake by the illuminated tumor as well as by the control tumor. If, indeed, circulating vasoactive mediators affect the control tumor, this systemic effect will likely induce subtle changes in the vascular system of other organs. To this end, PET of possible alternative target organs, such as the heart, could be attempted to reveal the existence of a systemic vasoconstriction effect induced by localized photodynamic activity.
Pharmacokinetic Modeling
The aim of this study was to demonstrate the feasibility of real-time imaging of transient metabolic processes during PDT. With such an aim, this pilot study did not attempt quantitative modeling of 18F-FDG uptake. The continuous-infusion protocol was designed, in the first place, to circumvent the limitations of multiple bolus injections of 18F-FDG, as were previously used to follow PDT kinetics (17). Deriving quantitative values from the observed changes in perfusion and metabolic activities will require the design and validation of a proper kinetic model, which will be addressed in future studies.
Future Perspectives
Even though these real-time 18F-FDG PET studies leave many unanswered questions on complex PDT tumor response mechanisms, the dissimilar transient metabolic processes revealed by changes in 18F-FDG uptake rates and response delays are striking evidence of different modes of action. The dramatic differences in 18F-FDG uptake profiles provide a rapid, real-time way to distinguish between direct and indirect mechanisms of tumor cell destruction and to assist in characterizing the photosensitizer and the PDT protocol. This is in sharp contrast to conventional, visual tumor response follow-up procedures that usually require many weeks of observation (40). Obviously, 18F-FDG PET can also be used to follow tumor regression after PDT at later intervals (17). However, the clear advantage of continuous 18F-FDG infusion and dynamic PET over other methods of assessing tumor response lies in its potential to reveal tumor response in real time, enabling the sequence of subtle transient metabolic processes within tumor tissues to be observed over time. In addition to identifying differences in mechanisms of action between various drugs, this technique allows for the rapid assessment of PDT protocols in order to optimize drug/light doses and their timing. In progress in our laboratory are further studies to expand real-time PET monitoring of tumor response to PDT by including radiotracers for blood flow, cell proliferation, hypoxia, and apoptosis. The same method could be considered to investigate the early response of tumors to other therapeutic approaches, such as chemotherapy or radiotherapy.
CONCLUSION
We have demonstrated the feasibility of using continuous 18F-FDG infusion and dynamic PET to study tumor metabolic response in real time during PDT in a small-animal model. This use proved particularly applicable for investigating tumoral and systemic transient metabolic changes resulting from PDT treatment and for characterizing the mechanisms of action of different photosensitizing drugs. Such dynamic small-animal PET can also be used as a rapid screening procedure for selecting new drugs and for optimizing treatment protocols. Because tumor response to PDT involves both a metabolic and a vasorestrictive component, we are currently expanding the procedure by including a blood-flow PET tracer in our PDT PET studies. The concept that has been demonstrated here—dynamic PET combined with a constant radiotracer infusion—can be applied to the real-time monitoring of other transient cellular and molecular processes, such as cell proliferation, protein synthesis, hypoxia, and apoptosis during and immediately after therapeutic intervention.
Acknowledgments
This work was supported by the Canadian Institutes of Health Research (CIHR grants MOP-44065 and MOP-15348). The authors would like to acknowledge the contribution of David Lapointe, who originally suggested the continuous infusion method used in this work to monitor tumor metabolic response in real time during PDT.
Footnotes
-
COPYRIGHT © 2006 by the Society of Nuclear Medicine, Inc.
References
- Received for publication September 2, 2005.
- Accepted for publication December 1, 2005.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}