


Abstract
Overexpression of multidrug resistance (MDR1) P-glycoprotein (Pgp) remains an important barrier to successful chemotherapy in cancer patients and impacts the pharmacokinetics of many important drugs, thus evoking a need to noninvasively interrogate Pgp transport activity in vivo. Methods: Cell tracer transport experiments as well as mouse biodistribution and microPET imaging studies were performed to characterize a nonmetabolized gallium(III) complex, gallium(III)-(bis(3-ethoxy-2-hydroxy-benzylidene)-N,N′-bis(2,2-dimethyl-3-amino-propyl)ethylenediamine) (Ga-[3-ethoxy-ENBDMPI])+, as a candidate SPECT (67Ga) and generator-produced PET (68Ga) radiopharmaceutical recognized by MDR1 Pgp. Results: The 67Ga-complex showed high membrane potential–dependent accumulation in drug-sensitive KB3-1 cells and modulator-reversible low accumulation in MDR KB8-5 cells. In KB8-5 cells, the median effective concentrations (EC50) of MDR modulators LY335979, PSC 833, and cyclosporin A were 69 nmol/L, 1 μmol/L, and 3 μmol/L, respectively. Using a variety of cells stably expressing MDR1 Pgp, multidrug resistance–associated proteins (MRP1–MRP6), or the breast cancer resistance protein (BCRP/MXR), the 67Ga-complex was shown to be readily transported by MDR1 Pgp and, to a much lesser extent, by MRP1, but not MRP2–MRP6 or BCRP/MXR. In a nude mouse xenograft tumor model, the 67Ga-complex produced a readily detected 3-fold difference between Pgp-expressing tumors and drug-sensitive tumors in the opposite flank. In mdr1a/1b(−/−) gene–deleted mice, the 67Ga-complex showed 17-fold greater brain uptake and retention compared with wild-type mice with no net difference in blood pharmacokinetics, consistent with transport in vivo by Pgp expressed at the capillary blood–brain barrier. This could be readily observed with microPET using the 68Ga-complex. Incidentally, wild-type mice showed heart-to-blood ratios of >100 by 1 h after injection and heart-to-liver ratios of 2.2 by 120 min. Conclusion: Molecular imaging of the functional transport activity of MDR1 Pgp with (67/68Ga-[3-ethoxy-ENBDMPI])+ may enable noninvasive SPECT/PET monitoring of the blood–brain barrier, chemotherapeutic regimens, and MDR1 gene therapy protocols in vivo. These Pgp-directed properties of the radiopharmaceutical may also translate favorably to myocardial perfusion imaging.
- MDR1 P-glycoprotein
- multidrug resistance
- blood–brain barrier
- perfusion imaging
- 67Ga/68Ga radiopharmaceutical
One of the best-characterized pathways for chemotherapeutic failure in malignancy are transport-mediated multidrug resistance mechanisms due to overexpression of the adenosine triphosphate–binding-cassette (ABC) superfamily of transporters (1). Biochemical and clinical data implicate P-glycoprotein (Pgp; ABCB1), a 170-kDa plasma membrane protein product of the MDR1 gene (1,2), the multidrug resistance–associated protein (MRP1; ABCC1), a related 190-kDa transporter protein (3,4), along with other selected members of the MRP subfamily (MRP1–MRP9; ABCC1–ABCC9) in drug resistance. Pgp confers resistance by reducing tumor cell content of a variety of structurally and functionally unrelated xenobiotics that are generally cationic through efflux transport, whereas MRPs (MRP1–MRP3) are thought to efflux transport conjugated or unconjugated generally anionic compounds as well as selected neutral hydrophobic chemotherapeutic agents. In addition, the breast cancer resistance protein (BCRP/MXR; ABCG2), an ABC “half transporter,” is also known to confer resistance to mitoxantrone and anthracyclines (5).
MDR1 Pgp has been a target for cancer therapy on several fronts. First, reversal of multidrug resistance in tumor cells by nontoxic agents that block the transport activity of Pgp has been an important target for pharmaceutical development (1). When coadministered with a cytotoxic agent, these inhibitors, known as MDR modulators, enhance net accumulation of cytotoxic compounds within the tumor cells. Second, transgenic expression of the MDR1 gene has been explored for hematopoietic cell protection in the context of cancer chemotherapy (6), wherein Pgp could protect hematopoietic progenitor cells from chemotherapy-induced myelotoxicity. In addition, MDR1 Pgp and MRP transporters normally are present in a variety of tissues, including intestinal epithelium, choroid plexus epithelium, testes, kidney, liver, adrenal, placenta, as well as capillary endothelial cells of the blood–brain barrier (BBB) (4,7,8). Thus, ABC transporters impact the pharmacokinetics of many common pharmaceuticals, and Pgp modulation is currently under evaluation as a means to improve oral absorption, for example, of chemotherapeutics and HIV-1 protease inhibitors (9,10). Therefore, a noninvasive method for determining Pgp-mediated drug transport activity could be important in predicting oral absorptivity, pharmacokinetics, and penetrance of MDR drugs into tumors, brain, germ cells, and the fetus.
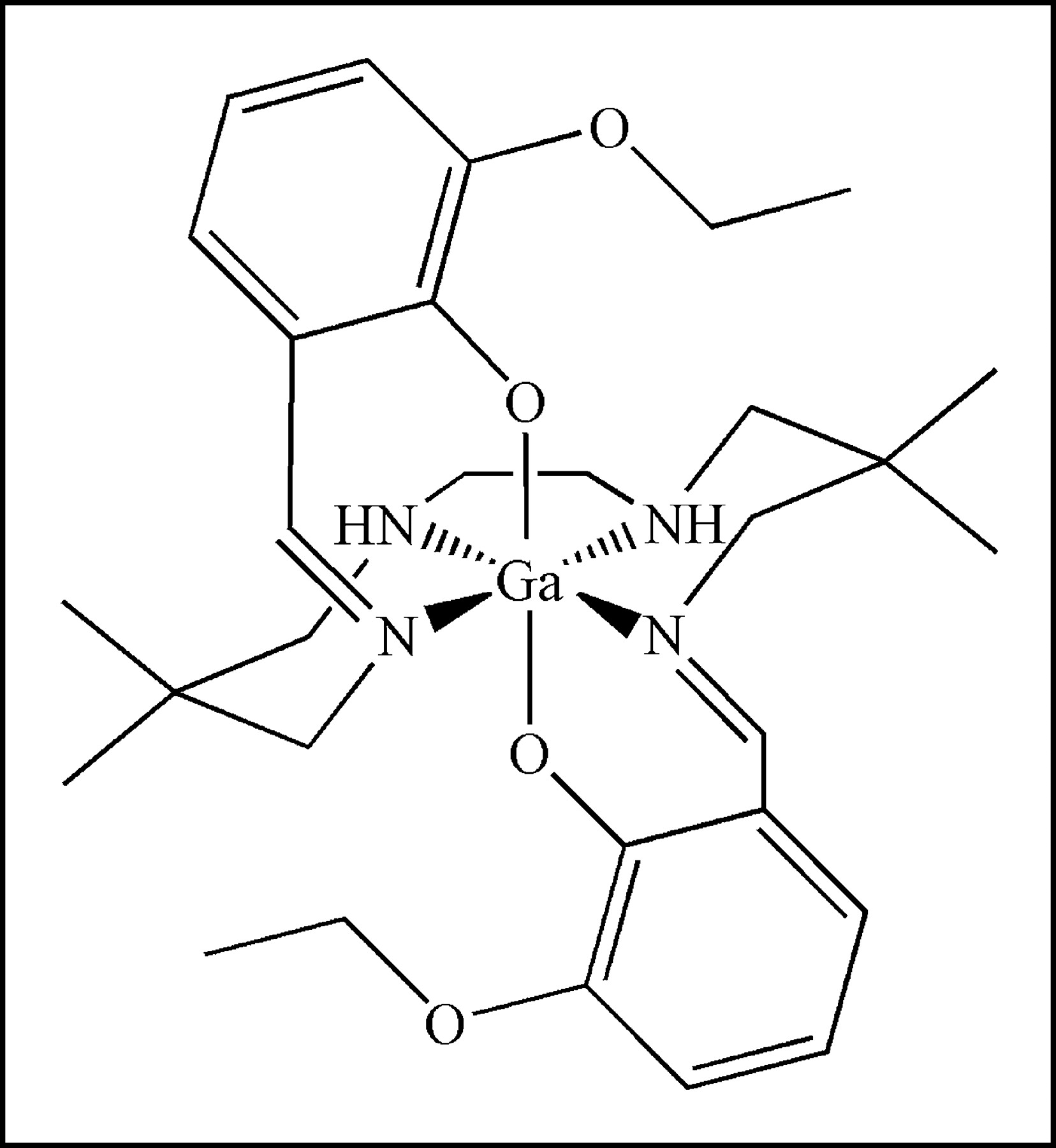
In this regard, various SPECT radiopharmaceuticals, exemplified by 99mTc-sestamibi, 99mTc-tetrofosmin, 99mTc-furifosmin, 99mTc-Q-58, and 99mTc-COMIBI (99mTc-tricarbonyl-tris(2-methoxyisobutyl-isonitrile)) (11,12) have been validated as transport substrates for MDR1 Pgp, enabling noninvasive imaging of Pgp-mediated transport activity in vivo. However, to exploit the greater sensitivity and quantification capabilities of PET, radiopharmaceuticals of short half-life (t1/2) isotopes have been sought. Based on structures of known MDR cytotoxic drugs or classic modulators, several 11C-labeled PET agents, including 11C-colchicine, 11C-verapamil, 11C-daunomycin, and 11C-paclitaxel have been reported (13–17). In addition, 18F-paclitaxel has been synthesized and evaluated in nonhuman primates (18,19). While promising data have been generated, some PET agents suffer from modest radiochemical yields and others from complex pharmacokinetics in vivo mediated, at least in part, by rapid metabolism of the radiolabeled compounds. Earlier, we reported the synthesis and crystal structure of a candidate Ga(III) complex targeting Pgp (20). Herein, we report biochemical, pharmacologic, tissue biodistribution, and brain microPET analysis of this novel radiopharmaceutical with potential applications in both SPECT (67Ga; t1/2 = 72 h) and PET (68Ga; t1/2 = 68 min).
MATERIALS AND METHODS
Solutions and Reagents
All reagents, except as indicated, were obtained from Sigma-Aldrich Chemical Co. Stock solutions of LY335979 (gift of Eli Lilly and Co.), PSC 833 (gift of Mallinckrodt, Inc.), methotrexate, and cisplatin were prepared in dimethyl sulfoxide (DMSO). The final concentration of DMSO in experimental buffers was <0.5%. Cyclosporin A was purchased as the Cremophor formulation and diluted in saline. The final concentration of Cremophor was <0.1%.
Control solution for transport experiments was a modified Earle’s balanced salt solution (MEBSS) containing (mmol/L): 145 Na+, 5.4 K+, 1.2 Ca2+, 0.8 Mg2+, 152 Cl−, 0.8 H2PO4−, 0.8 SO42−, 5.6 dextrose, 4.0 N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid), and 1% bovine calf serum (v/v), pH 7.4 ± 0.05. Dulbecco’s modified Eagle medium with 1% bovine calf serum was used as a control solution for transport experiments with incubation times of >90 min.
Cell Culture
Monolayers of drug-sensitive human epidermoid carcinoma KB3-1 cells and the colchicine-selected KB8-5 and KB8-5-11 MDR cell lines were grown as previously described (21,22). Growth conditions for stably transfected MCF-7 and MCF-7/MDR1 cells, H69 and drug-selected H69AR cells, as well as stably transfected LLC/CMV and LLC/cMoat-1 cells, NIH3T3 and 3T3/MRP4 cells, and HEK293 and 293/MRP3, 293/MRP5 and 293/MRP6 cells have been described (22–31). Parental S1M1 and MCF-7 cells and their drug-selected derivative cells, S1M1-80 and MCF-7-AdVp3000, respectively (gift of Dr. Susan Bates), were maintained as described (5,27). ABC transporter status of all cell lines is summarized in Table 1.
Summary of Cell Lines
Preparation of ([3-ethoxy-ENBDMPI]Ga)+ ClO4− (1a)
The synthesis of 1a has been described earlier (20). Briefly, a heptadentate precursor ligand, 2-(2-hydroxy-3-ethoxyphenyl)-1,3-bis[4-aza-5-(2′-hydroxy-3′-ethoxyphenyl)-2″,2‴-dimethyl-but-4′-ene-1′-yl]-1,3-imidazolidine (0.14 g, 0.21 mmol), and gallium(III)acetylacetonate (0.08 g, 0.21 mmol) were reacted in methanol for 30 min, followed by addition of potassium perchlorate (0.03 g, 0.21 mmol) dissolved in 0.5 mL of hot water. Slow evaporation of methanol yielded pale yellow needles that were washed with methanol, then washed with ether, and dried to yield 1a (0.10 g, 0.14 mmol, 67.6%).
Preparation of 67/68Ga(III) complexes (1b)
Synthesis of the radiolabeled gallium(III) complex was described earlier (20). 67Ga was obtained as the commercial citrate salt in water (Mallinckrodt Inc.), whereas 68Ga was obtained as GaCl3 after elution of a Ge/Ga generator (Dupont) with 6N HCl. Both were converted to the respective 67/68Ga-(acetylacetonate)3 by reacting with acetylacetone using standard procedures (32). Radiolabeled 67Ga- and 68Ga-complexes were obtained through addition of 67Ga- or 68Ga-(acetylacetonate)3 to heptadentate Schiff-base precursor ligand in ethanol at 80°C for 30 min. The contents were cooled, diluted with saline to 10% ethanol, and passed through a nylon syringe filter. The desired product was purified on a C18 silica cartridge (Sep-Pak), eluted with saline:ethanol (90:10), assessed for radiopharmaceutical purity by radio–thin-layer chromatography (radio-TLC) on C18 plates (Whatman) using an eluent mixture of methanol:saline (90:10; Rf: 0.47), and found to be ∼100% pure.
Cell Transport Studies
Coverslips with confluent cells were used for studies of cell transport and tracer kinetics as previously described (33). Cells on coverslips were removed from culture media and washed for 15–30 s in control solution, then immersed in 60-mm glass or plastic Petri dishes containing 4 mL of loading solution consisting of MEBSS control solution, drug, or vehicle, and 67Ga-complex 1b was diluted to the desired concentration (8–24 pmol/MBq; 37–74 kBq/mL) (<1% ethanol final). Coverslips with cells were removed after 90 min or at various times for kinetic studies, rinsed 3 times in 25 mL ice-cold control solution for 8 s each to clear extracellular spaces, and placed in 35-mm plastic Petri dishes. For washout studies, cells were first equilibrated in loading solution for 90 min, then washed 3 times in ice-cold control solution, and thereafter incubated in control solution (37°C) for various washout times. Subsequently, cells were again rinsed 3 times in ice-cold control solution and placed in 35-mm plastic Petri dishes. In both protocols, cells were then extracted in 1% sodium dodecyl sulfate with 10 mmol/L sodium borate before protein assay by the bicinchoninic acid analysis (Pierce Chemical Co.) method, using bovine serum albumin as the protein standard. Aliquots of the loading solution and 67Ga-complex 1b stock solutions also were obtained for standardizing cellular data with the extracellular concentration of 1b. For cells in suspension culture, transport studies were performed as described (24,27). All cell extracts, 67Ga-complex 1b stock solutions, and loading solution samples were assayed for γ-activity in a well-type sodium iodide γ-counter (Cobra II; Packard). The absolute concentration of total Ga-complex 1b in solution was determined from the activity of stock solutions and the specific activity of Ga based on the manufacturer’s specifications for 67Ga. Data are reported as fmol Ga-complex 1b (mg protein)−1 (nmol/L)−1 as previously described (20), with (nmol/L) representing the total concentration of Ga-complex in the extracellular buffer.
Western Blots
Pgp was detected in crude, enriched membrane preparations from cell lines using Western blotting with monoclonal antibody (mAb) C219 (27). Immune complexes were identified with sheep antimouse antibody (1:2,000 dilution) coupled to horseradish peroxidase using the ECL Western blotting detection system (Amersham Life Sciences).
Biodistribution Studies
All animal procedures were approved by the Washington University Animal Studies Committee. Distribution of 67Ga-complex 1b in tissues of male 8-wk-old FVB wild-type (WT) and mdr1a/1b(−/−) gene-deleted (KO) mice (Taconic) was determined as previously described (22,34). For tumor xenografts, 7- to 8-wk-old male NCr athymic nu/nu mice (Taconic) were inoculated subcutaneously with 2 × 106 drug-sensitive KB3-1 cells in the left flank and drug-resistant KB8-5-11 cells in the right flank, and tumors were allowed to grow as described previously (21). 67Ga-Complex dissolved in saline:ethanol (90:10) was diluted in saline for a final concentration of 740 kBq/mL. All mice were anesthetized by metofane inhalation and injected with 74 kBq of radiotracer via bolus injection through a tail vein. Animals were sacrificed by cervical dislocation at 5, 15, 60, and 120 min after injection (n = 2 or 3 each). Blood samples were obtained by cardiac puncture, organs and tumors were then harvested rapidly, and all tissue samples were analyzed for γ-activity.
For analysis of metabolites, mouse tissues harvested 15 min after injection were minced in ethanol:saline (90:10), transferred to conical tubes, and centrifuged. Supernatants, containing the majority of the counts, were removed and analyzed through radio-TLC, revealing ∼100% parental complex. Data are expressed as the percentage injected dose (%ID) per gram of tissue ([tissue kBq] [injected kBq]−1 [g tissue]−1 × 100) or the percentage injected dose per organ ([tissue kBq] [injected kBq]−1 × 100). While the former are presented herein, please contact the corresponding author for the latter as supplemental tables.
Imaging Studies
Approximately 24-wk-old WT and KO mice were anesthetized with isoflurane inhalation. 68Ga-Complex (7.4 MBq in 100 μL 10% ethanol/saline) was injected via a tail vein into mice positioned in a microPET R4 detector (Concorde Microsystems) and imaged (10-min acquisition time; 1 bed position; filtered-backprojection reconstruction; isotropic image resolution, 1.8 mm) (35). MicroPET images were corrected for decay, but not attenuation.
Analysis
All data points for transport assays were determined from preparations obtained from the same culture. EC50 values (half-maximal effective concentration of drug) for modulators were estimated by a computer fit (σ-Plot; Jandel Scientific) of concentration–effect curves of Ga-complex transport inhibition as described (27,36). From biodistribution time–activity curves, the area under the curve (AUC) was calculated using trapezoidal integration (KaleidaGraph; Synergy Software) and reported as tissue kBq (injected kBq)−1 (g tissue)−1 × 100 × minute. Data are generally reported as mean ± SEM. Pairs were compared by the Student t test. P values ≤ 0.05 were considered significant.
RESULTS
Characterization of (Ga-[3-Ethoxy-ENBDMPI])+
Both 67Ga, a single-photon radioisotope, and 68Ga, a generator-produced positron-emitting radioisotope, were used for radiosynthesis of (67/68Ga-[3-ethoxy-ENBDMPI])+ complex 1b (Fig. 1). Both 67Ga- and 68Ga-complexes were obtained in 35% radiochemical yield and ∼100% radiochemical purity. The total time required for reaction and work-up was compatible with that of short-lived isotopes. The longer-lived 67Ga-complex 1b was tested and found to be stable in human serum at 37°C for up to 24 h. Furthermore, radio-TLC analysis of tissue extracts 15 min after intravenous injection of 67Ga-complex 1b in mice, a time corresponding to initial imaging of tumors and the BBB, demonstrated the existence of only nonmetabolized parental compound in heart, liver, and kidney.

Structure of ([3-ethoxy-ENBDMPI]Ga)+ (1), a MDR1 Pgp-targeted gallium(III) Schiff-base complex. In the text, 1a represents unlabeled Ga-complex and 1b represents 67Ga- or 68Ga-complexes.
Cellular Transport Kinetics
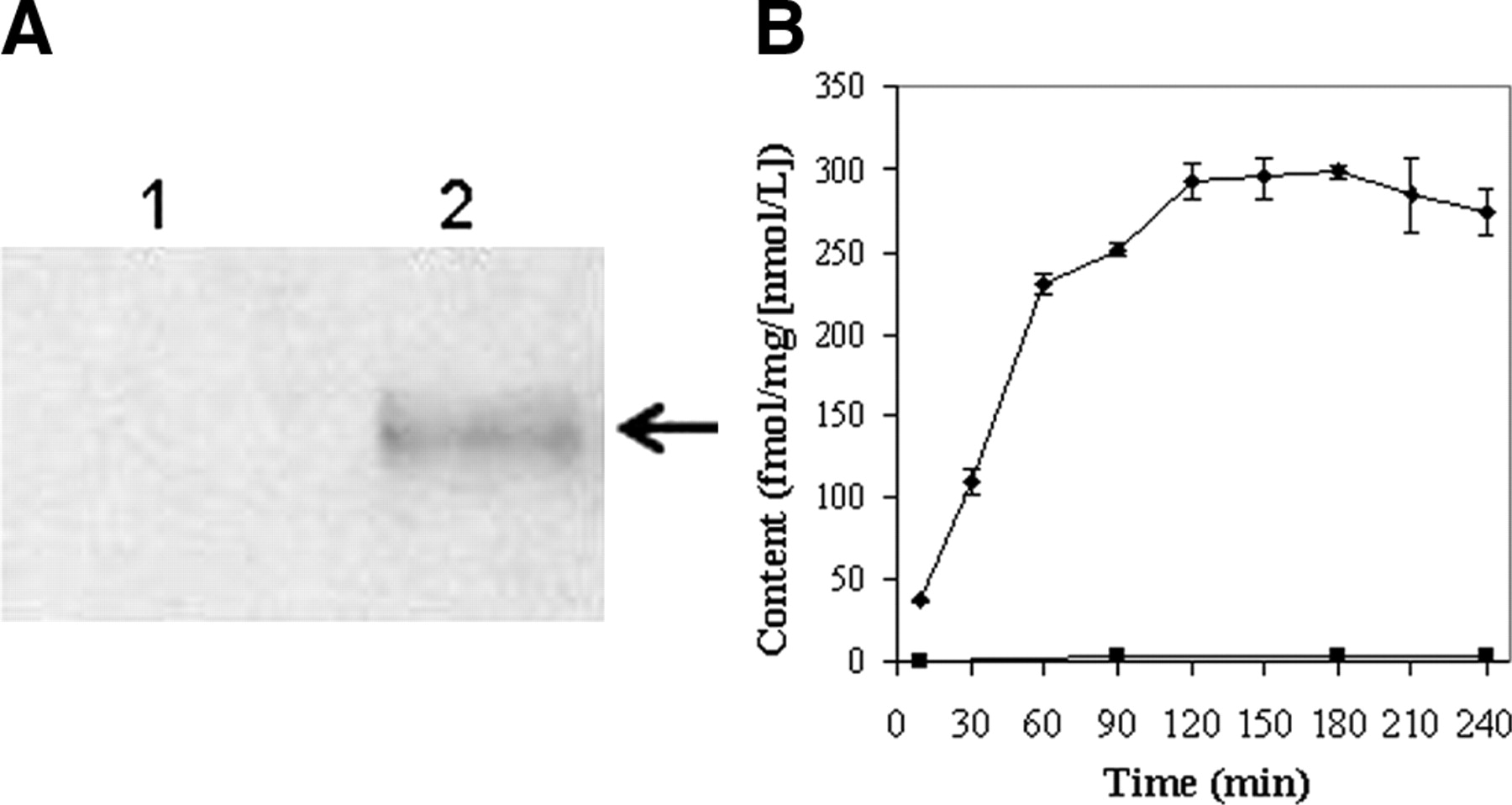
KB8-5 cells express modest levels of Pgp as documented by Western blot analysis with mAb C219 (Fig. 2A); therefore, MDR1 Pgp-mediated efflux transport would be expected to result in a decreased net cellular accumulation in KB8-5 cells compared with that of the parental KB3-1 cells. In KB3-1 cells, 67Ga-complex 1b approaches a plateau within 90 min (Fig. 2B). The steady-state value for the 67Ga-complex was 292 ± 10 fmol (mg protein)−1 (nmol/L)−1. Given a typical cell water space of 4 μL (mg protein)−1 (37), this indicated an intracellular-to-extracellular ratio of ∼75, consistent with concentrative accumulation of the radiotracer within Pgp-negative cells. In contrast, in Pgp-positive KB8-5 cells, steady-state accumulation of the radiotracer was low, resulting in a KB3-1–to–KB8-5 uptake ratio of 104, a functional measure of high interaction of this compound with Pgp. In addition, cellular accumulation of 67Ga-complex 1b in KB3-1 cells at 4°C was minimal over 2 h (1.6 ± 0.2 fmol [mg protein]−1 [nmol/L]−1; n = 4), indicating a strong temperature-dependent process for permeation of the radiopharmaceutical. Furthermore, in KB8-5 cells, as expected, washout kinetics of the 67Ga-complex at 37°C demonstrated a 10-fold faster rate (k = 0.20 min−1) compared with that of KB3-1 cells (k = 0.02 min−1) (Fig. 3). However, the overall clearance from KB3-1 cells was slower compared with that of other Pgp-targeted radiopharmaceuticals such as 99mTc-sestamibi (27,33). Zero-trans washout was only ∼85% complete by 2 h (Fig. 3), consistent with the modestly higher octanol:water partitioning coefficient for Ga-complex 1b (log P = 1.7) versus Tc-sestamibi (log P = 1.1). Therefore, residual activity retained within intracellular compartments, even after 2 h, may be due to nonspecific interaction of 1b with hydrophobic (i.e., membrane) compartments within cells in the absence of Pgp.
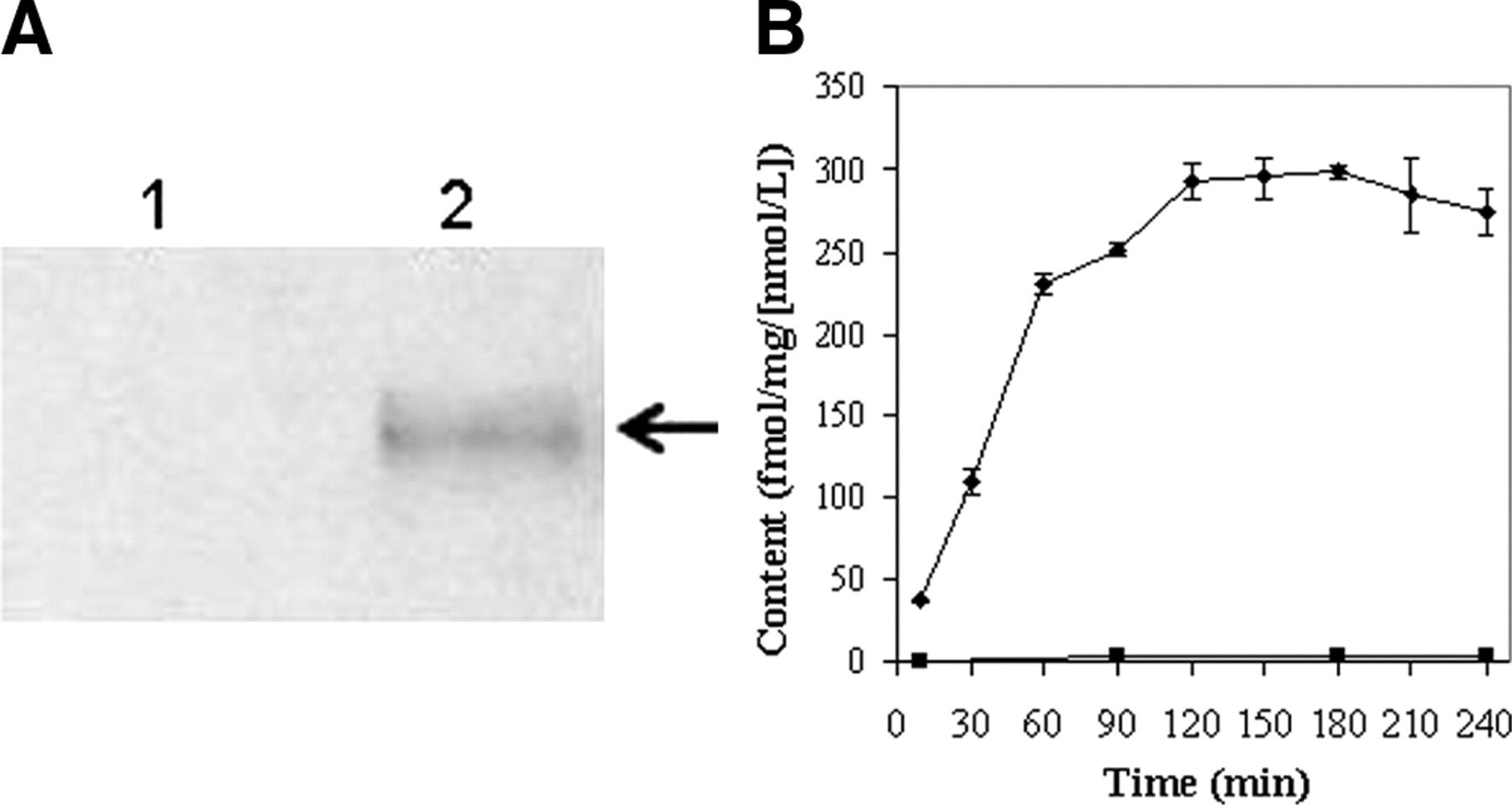
(A) Western blot (anti-Pgp mAb) of KB3-1 (lane 1) and KB8-5 (lane 2) cell membranes. Arrow demarcates Pgp. (B) Kinetics of 67Ga-complex 1b accumulation into drug-sensitive KB3-1 (♦) and Pgp-expressing KB8-5 (▪) tumor cells. Cells were incubated in MEBSS loading buffer for the indicated times, then washed, and cell-associated activity was determined. Each point represents mean of 3 or 4 determinations; bars represent ±SEM when larger than the symbol. Plot is representative of 2 independent experiments.
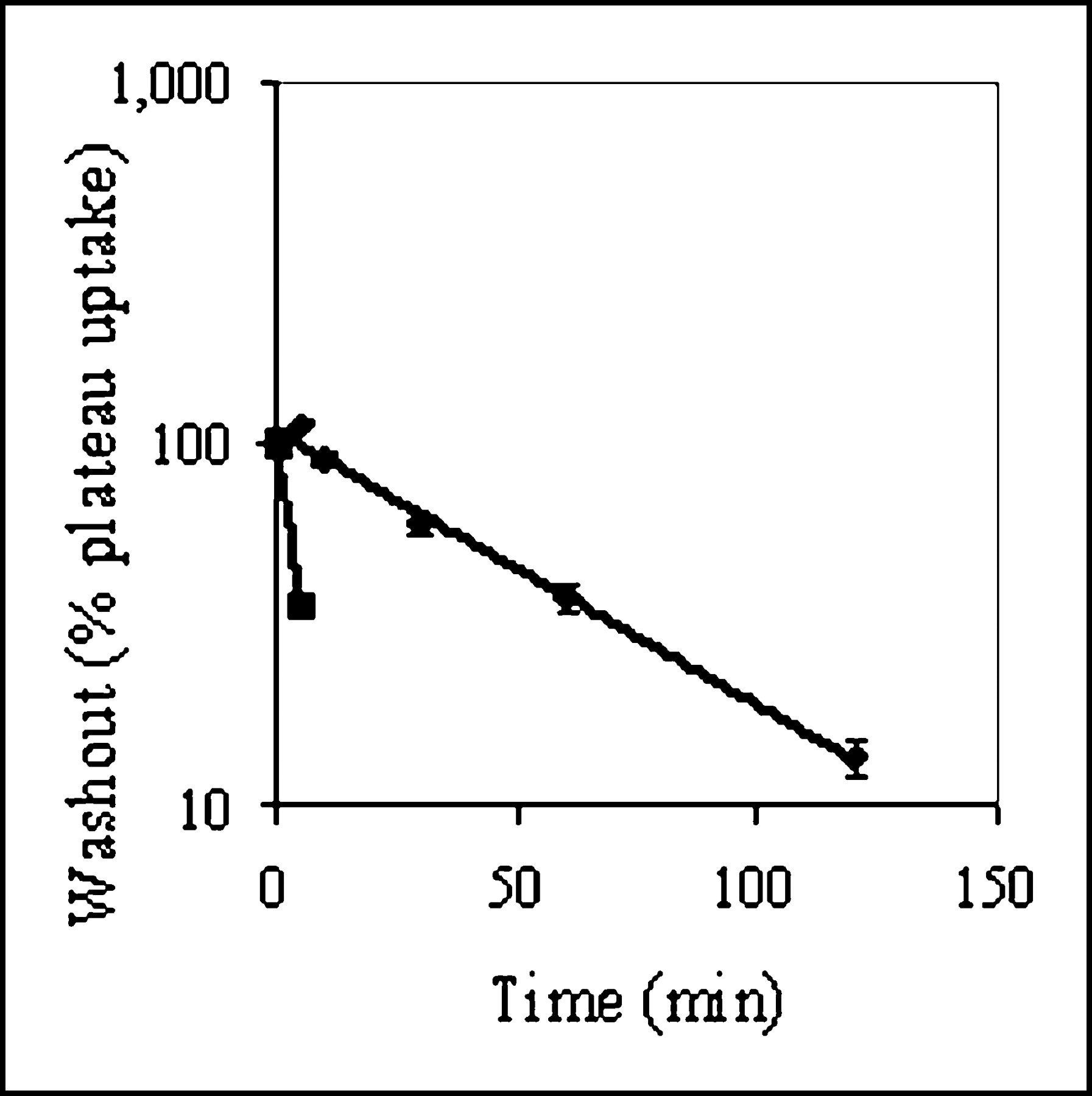
Washout kinetics of 67Ga-complex 1b from KB3-1 (♦) and KB8-5 (▪) cells at 37°C. Cells were preequilibrated in MEBSS loading buffer (37°C) for 90 min and then transferred to isotope-free buffer. Cell-associated activity as a function of time in washout buffer is shown expressed as percentage of plateau uptake values. Each point represents mean of 3 or 4 determinations; bars represent ±SEM when larger than the symbol. Note logarithmic scale for y-axis. Solid line represents a curve fit with a single exponential equation (♦, k = 0.02; ▪, k = 0.20).
To further evaluate the mechanism(s) involved in intracellular accumulation of this tracer, KB3-1 cells were incubated with the 67Ga-complex in buffer containing 130 mmol/L K+/20 mmol/L Cl− and the potassium ionophore valinomycin (1 μg/mL). Under these conditions, electrical potentials of the mitochondrial membrane (ΔΨ) and plasma membrane (Em) are depolarized to zero, thereby eliminating the inward driving force for hydrophobic cationic compounds such as 1b. Thus, cellular accumulation is expected to decrease, and any net uptake (in-to-out ratio > 1) under these conditions is indicative of nonspecific membrane absorption. Under zero potential conditions, cell tracer accumulation decreased to 185 ± 5.0 fmol (mg protein)−1 (nmol/L)−1 compared with a control value of 372 ± 13 fmol (mg protein)−1 (nmol/L)−1 for this experiment, indicating a significant role for membrane potential in localization of the radiotracer as previously described at an earlier time point (20). However, the residual potential-independent retention was equivalent to an apparent in-to-out ratio of 46, indicating, in the absence of Pgp, a substantial level of nonspecific interactions with cellular constituents. Expression of Pgp in KB8-5 cells effectively countered both potential-independent as well as potential-dependent uptake components.
Effect of MDR1 Pgp Modulators on Tracer Accumulation in MDR Cells
Further validation of 67Ga-complex 1b as a substrate for MDR1 Pgp was provided by monitoring tracer accumulation in KB8-5 cells in the presence of known Pgp inhibitors. Inhibition of Pgp-mediated transport of the 67Ga-complex would be indicated by enhanced net accumulation of the radiotracer. The Pgp inhibitors cyclosporin A (a cyclic undecapeptide), LY335979 (a difluorocyclopropyl dibenzosuberane (38)), and PSC 833 (a cyclic analog of cyclosporin A (39)) enhanced plateau (90 min) tracer accumulation in KB8-5 cells (Fig. 4). LY335979 was most potent (EC50 = 69 nmol/L), cyclosporin was least potent (EC50 = 3 μmol/L), and PSC 833 demonstrated intermediate potency (EC50 = 1 μmol/L), consistent with the known potency rank order of these MDR inhibitors (27). Different maximal efficacies were observed at concentrations beyond the customary pharmacologic concentrations for the drugs. As expected, drugs such as methotrexate and cisplatin (100 μmol/L), not belonging to the MDR phenotype, did not enhance cell tracer accumulation (data not shown). Furthermore, in KB8-5 cells, the respiratory uncoupler carbonyl cyanide m-chlorophenylhydrazone (CCCP), a reagent known to depolarize mitochondrial membrane potential (33), abrogated the LY335979-induced enhancement of the 67Ga-complex (control uptake, 3.8 ± 0.4 fmol [mg protein]−1 [nmol/L]−1; + CCCP [5 μmol/L], 2.9 ± 0.2; + LY335979 [1 μmol/L], 312.0 ± 8.2; + CCCP + LY335979, 50.2 ± 1.9), indicating that inhibition of Pgp with LY335979 allowed the radiopharmaceutical to concentrate in mitochondria in a potential-dependent manner.

Concentration–effect curves of Pgp inhibitors LY335979 (▪), cyclosporin A (▴), and PSC 833 (♦) on cellular accumulation of 67Ga-complex 1b in MDR KB8-5 cells. Cells were incubated for 90 min in loading buffer containing tracer and various concentrations of the indicated inhibitor. Each point represents mean of 4 determinations; bars represent ±SEM when larger than the symbol.
Evaluation of 67Ga-Complex 1b in MCF-7 Cells Transfected with MDR1 Pgp
Cell lines derived from stepwise selection with MDR drugs, such as KB8-5, may coexpress multiple mechanisms of drug resistance, thereby complicating interpretation of results (40). Thus, we used nondrug–selected MCF-7 cells stably transfected with MDR1 (29) for confirmation of 67Ga-complex transport. Control MCF-7 cells lack immunodetectable Pgp, whereas MCF-7/MDR1 cells express readily detected Pgp by Western blotting analysis (29). Therefore, these matched cell lines provided a robust model system for analysis of MDR1 Pgp without confounding effects of prior exposure to MDR drugs. Compared with parental cells, steady-state accumulation of 67Ga-complex 1b was almost 40-fold less in MCF-7/MDR1 cells (155 ± 6.2 fmol [mg P]−1 [nmol/L]−1 vs. 3.8 ± 0.7 fmol [mg P]−1 [nmol/L]−1, respectively) (Fig. 5). LY335979 enhanced accumulation of the radiotracer in MCF-7/MDR1 cells, consistent with blockade of Pgp-mediated efflux, whereas LY335979 showed a minimal effect in MCF-7 cells.
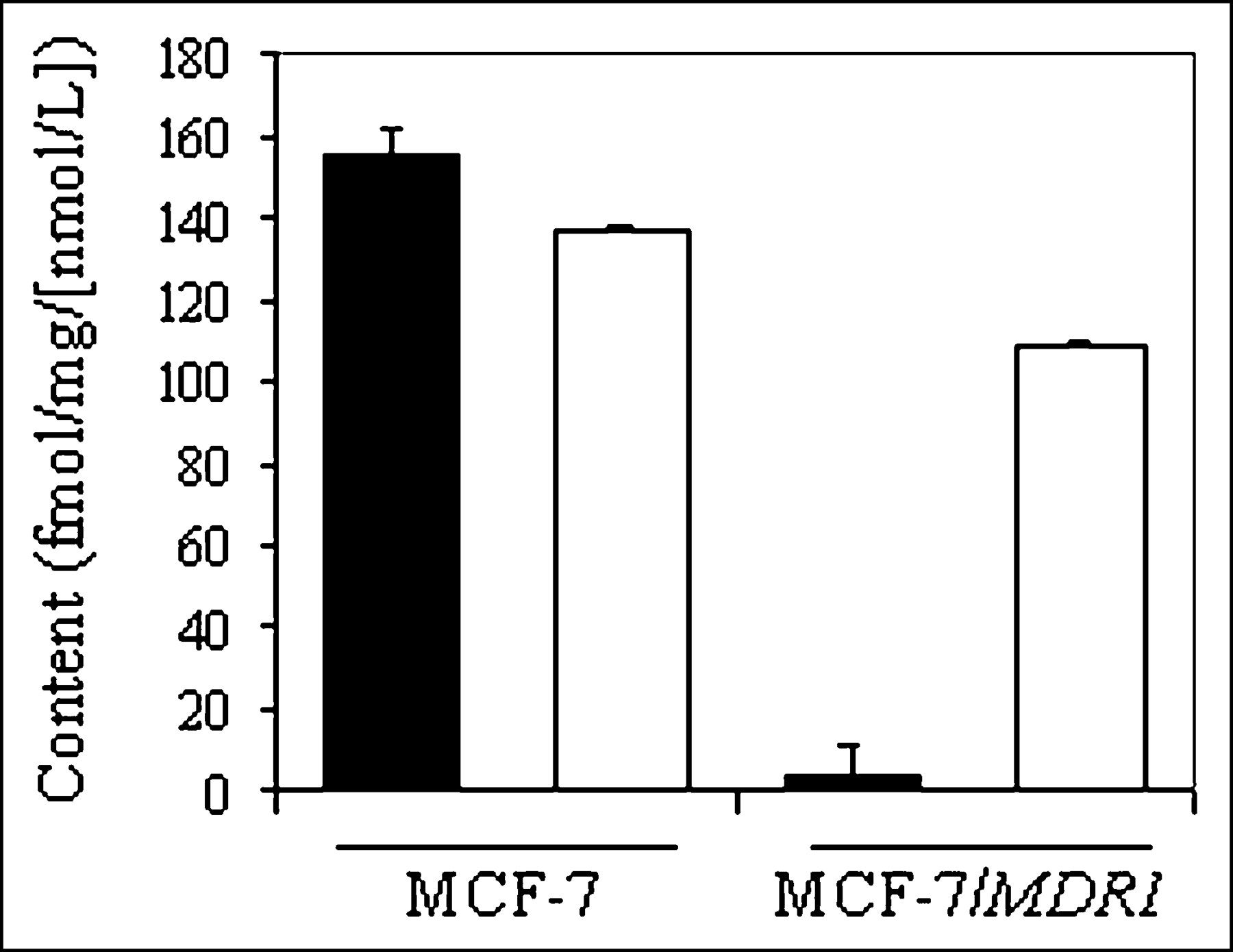
Cellular accumulation (90 min) of 67Ga-complex 1b in control MCF-7 and MCF-7/MDR1 cells in absence (solid bar) and presence (open bar) of MDR modulator LY335979 (1 μmol/L). Each bar represents mean of 4 determinations; small bars represent SEM.
Cross-Reactivity of 67Ga-Complex 1b with Other Transporters
To further evaluate the sensitivity and selectivity of the 67Ga-complex to report expression of MDR1 Pgp and to monitor cross-reactivity with other ABC superfamily members, cell accumulation assays were performed in cell lines that express variable levels of MDR1 Pgp and human multidrug resistance–associated proteins (MRP1–MRP6).
Cellular accumulation of radiotracer over 90 min in control H69 and H69AR cells was found to be 968 ± 57 and 103 ± 2.1 fmol (mg protein)−1 (nmol/L)−1, respectively (Fig. 6A), indicating a 9-fold decrease in accumulation of the tracer due to overexpression of MRP1 in H69AR cells. In addition, in H69 cells, depolarization of membrane potential with 130 mmol/L K+/20 mmol/L Cl− buffer plus valinomycin (1 μg/mL) decreased tracer content to 203 ± 7.0 fmol (mg protein)−1 (nmol/L)−1, indicating a substantial nonspecific in-to-out uptake ratio of 51, similar to that observed in KB cells. The Pgp inhibitor LY335979 showed no significant effect on cellular uptake of tracer in either control or H69AR cells. Furthermore, accumulation of the 67Ga-complex was monitored in cells stably transfected with human MRP2, MRP3, MRP4, MRP5, or MRP6 to evaluate the cross-reactivity with these other MRP homologs. LY335979 enhanced uptake in all of the various control cells and their MRP2–MRP6 transfectants (Fig. 6B), documenting the constitutive expression of endogenous Pgp in these cell lines. No further change in tracer content was observed by concurrent overexpression of human MRP2–MRP5, indicating a lack of cross-reactivity of the 67Ga-complex with these transporters. Overexpression of human MRP6 induced a slight increase in tracer content in the absence of LY335979 and an opposing 25% decrease in the presence of LY335979 (P < 0.02), which together were interpreted as biochemically insignificant. Overall, these results indicated that the radiotracer cross-reacted with only MRP1, but to a modest degree (cell uptake ratio = 9) compared with Pgp (cell uptake ratio = 104).
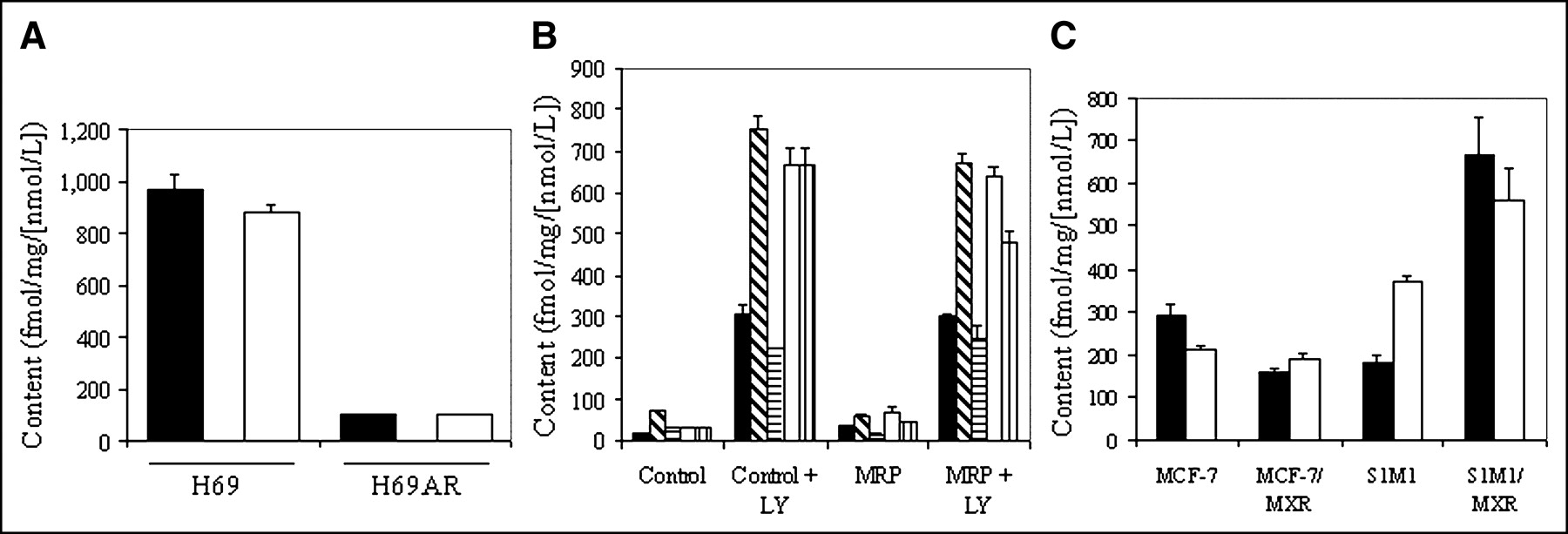
Effect of MRP family members on tracer uptake. (A) Cellular accumulation (90 min) of 67Ga-complex 1b in control H69 and MRP1-expressing H69AR cells in absence (solid bar) and presence (open bar) of MDR modulator LY335979 (1 μmol/L). (B) Cellular accumulation (90 min) of 67Ga-complex 1b in various parental control cells or matched cells expressing MRP2 (▪), MRP3 ( ), MRP4 (
), MRP4 ( ), MRP5 (□), or MRP6 (
), MRP5 (□), or MRP6 ( ) in absence or presence of MDR modulator LY335979 (1 μmol/L). (C) Cellular accumulation (90 min) of 67Ga-complex 1b in human breast carcinoma (MCF-7) and human colon carcinoma (S1M1) control cells compared with drug-selected cells expressing the half transporter BCRP/MXR in absence (solid bar) and presence (open bar) of MDR modulator LY335979 (1 μmol/L). Each bar represents mean of 3 or 4 determinations; small bars represent SEM.
) in absence or presence of MDR modulator LY335979 (1 μmol/L). (C) Cellular accumulation (90 min) of 67Ga-complex 1b in human breast carcinoma (MCF-7) and human colon carcinoma (S1M1) control cells compared with drug-selected cells expressing the half transporter BCRP/MXR in absence (solid bar) and presence (open bar) of MDR modulator LY335979 (1 μmol/L). Each bar represents mean of 3 or 4 determinations; small bars represent SEM.
Breast cancer resistance protein (BCRP/MXR) is an ABC half-transporter that is known to dimerize to form an active complex. Thus, to evaluate potential cross-reactivity with BCRP/MXR, 67Ga-complex 1b was evaluated in human breast carcinoma MCF-7 (control), MCF-7-AdVp3000 (+MXR), colon carcinoma S1M1 (control), and S1M1-80 (+MXR) cells (Fig. 6C). Uptake was found to be slightly lower in MCF-7/MXR cells and higher in S1M1/MXR cells compared with that of the paired parental cells. Furthermore, LY335979 (1 μmol/L) enhanced uptake of 67Ga-complex 1b in MCF-7/MXR cells and control S1M1 cells, whereas it reduced uptake in MCF-7 control cells and S1M1/MXR cells. The ≤2-fold MXR-induced transport differences in opposing directions for each cell type were considered together to be biochemically insignificant. Thus, 67Ga-complex 1b is not transported significantly by BCRP/MXR.
Probing Pgp Transport Activity in Vivo
We used the biodistribution of 67Ga-complex 1b to probe Pgp function in a nude mouse tumor xenograft model. Bilateral tumors were produced in opposite flanks of nu/nu mice with drug-sensitive KB3-1 cells and multidrug-resistant KB8-5-11 cells (21). The Pgp-expressing KB8-5-11 tumor accumulated less 67Ga-complex 1b compared with that of the parental tumor in the opposite flank (Table 2), producing a readily detected 3-fold difference by 1 h after injection. Heart, an organ rich in mitochondria, but lacking Pgp, showed a tissue-to-blood ratio of >100 by 1 h after injection, as expected for unopposed membrane potential–driven influx of the tracer.
Biodistribution (%ID/g) of 67Ga-Complex 1b in Tumor-Bearing Nude Mice
Drug-transporting Pgps (MDR1 in humans; mdr1a in mice) are expressed on luminal surfaces of brain endothelial cells and limit entry of amphipathic compounds into the central nervous system (41). Therefore, these proteins constitute an important component of the BBB. To demonstrate application of the 67Ga-complex as a functional marker of Pgp-mediated transport activity at the BBB, we determined the biodistribution of 67Ga-complex 1b in WT and mdr1a/1b(−/−) (KO) mice. Relative to WT mice, KO mice showed 10-fold more 67Ga-complex 1b in brain parenchyma 5 min after injection of the complex (Tables 3 and 4). Additionally, the AUC5–120 of 67Ga-complex 1b in KO brain was 17-fold greater than that of WT mice (P < 0.005) (Table 5). By contrast, the AUC5–120 of the radiotracer in the blood of KO mice was only 1.2-fold that of WT control mice (P > 0.5). Because blood flow to the brain does not differ between mdr1 KO mice and WT mice (42), differences in initial penetration and retention of radiotracer in the brain tissue of KO mice cannot be attributed to differences in cerebral perfusion and blood clearance.
Biodistribution (%ID/g) of 67Ga-Complex 1b in FVB WT Mice
Biodistribution (%ID/g) of 67Ga-Complex 1b in FVB mdr1a/1b(−/−) Mice
Biodistribution of 67Ga-Complex 1b in FVB WT and mdr1a/1b(−/−) Mice (AUC5–120; %ID/g × min)
In addition, the liver, a polarized tissue expressing Pgp, showed twice the retained activity (AUC5–120) in KO mice compared with that in WT mice, whereas the heart, a Pgp-negative tissue, did not show any significant difference (P > 0.5). The heart-to-liver ratio in WT mice increased to 2.2 by 120 min, whereas in KO mice it was 0.4 (Tables 3 and 4), a low value reflecting the minimal non-Pgp–mediated clearance of 67Ga-complex 1b from livers of KO mice.
To further evaluate the potential utility of the Ga-complex as a PET radiotracer in vivo, 68Ga-complex 1b was obtained by reaction of 68Ga-(acetylacetonate)3 with precursor ligand, purified, and injected via tail vein into mice. Representative microPET images of brain tracer uptake at 5 min after injection in WT and KO mice are shown in Figure 7. Increased penetration and retention of the PET radiotracer were apparent in the brain of KO mice compared with that of WT mice, consistent with the biodistribution data.

MicroPET image of 68Ga-complex 1b in brains of FVB WT and mdr1a/1b(−/−) mice. After injection of mice with an intravenous bolus of radiopharmaceutical, images of abdomen, thorax, and head were obtained with microPET scanner. Representative coronal images of WT (left) and mdr1a/1b(−/−) (right) mice obtained 5 min after injection are shown. A body outline is included for orientation.
DISCUSSION
Though well-characterized for contributing to drug resistance in cancer chemotherapy, MDR1 Pgp also has emerged as a major contributor to the bioavailability, BBB penetration, and pharmacokinetics of a wide variety of drugs, including HIV protease inhibitors, antidepressants, antiarrythmics, and new molecular therapeutics such as imatinib (Gleevec) (43,44). Therefore, agents that probe Pgp transport are desired to guide chemotherapeutic choices, drug regimens, and gene therapy strategies before treatment.
Various single-photon agents such as 99mTc-sestamibi and 99mTc-tetrofosmin, originally developed for myocardial perfusion imaging, have been shown to be robust substrates of MDR1 Pgp. These agents have been shown to detect transport activity of Pgp in normal human tissues (45), in human tumors (46,47), and in patients treated with MDR modulators that inhibit transport activity of Pgp (1,45,48,49). They can predict chemotherapeutic failure in breast cancer, lung cancer, lymphoma, and osteosarcoma (50–54). However, nonmetabolized PET radiopharmaceuticals designed to interact with MDR1 Pgp with high specificity would offer alternative probes with enhanced quantitative capacity for molecular imaging of Pgp activity in tissues in vivo.
In considering alternative radiolabels apart from conventional cyclotron-produced PET isotopes, generator-produced 68Ga may offer practical radiopharmacy and distribution advantages. Among Ga-labeled compounds, selected monocationic, hydrophobic Ga(III) complexes with octahedral geometry arising from an N4O2 donor core can diffuse into intracellular compartments and, in the absence of Pgp, concentrate as a result of inwardly directed electrochemical driving forces (20). Hydrophobicity and pharmacologic activity can be controlled by variation of the substituents on the periphery of the molecule without impacting the N4O2 coordination core. Earlier, we reported the synthesis and structure of (Ga-[3-ethoxy-ENBDMPI])+ (1b) (20). In the present study, we now show that this radiotracer is recognized by MDR1 Pgp as an efflux transport substrate with high sensitivity. Indeed, the high steady-state nontarget-to-target cellular accumulation ratios compared with 99mTc-sestamibi and 99mTc-tetrofosmin (27) demonstrated the superior sensitivity of 1b in detecting the outward transport activity of MDR1 Pgp.
Selected MRP family members also may impact resistance to cytotoxic drugs. Within the MRP superfamily, MRP1, -2, -3, and -6 are organic anion transporters, whereas MRP4 has been shown to confer resistance to antiviral acyclic nucleotide analogs (55). Similarly, MRP5 has been shown to act as an energy-dependent transporter of nucleotides (56). Drugs that are recognized by the MRP superfamily also include some neutral and cationic compounds (3). Ga-complex 1b demonstrated a modest degree of cross-reactivity with MRP1, but the sensitivity for detection of transport activity was low compared with that of MDR1 Pgp. By “modest,” we identified a 9-fold MRP1-dependent change in cell tracer content versus up to 100-fold for MDR1 Pgp. The ratio of MRP1-dependent to MDR1 Pgp-dependent transport is thus ∼0.1 with Ga-complex 1b, a ratio half that of 99mTc-sestamibi, another agent that also shows a modest cross-reactivity with MRP1 in cells in culture and a ratio of MRP1-dependent to Pgp-dependent transport of ∼0.2. However, in contrast to Pgp, an MRP1-dependent signal has not been observed with 99mTc-sestamibi in patients in several clinical trials attempting to define this. It would appear that the modest MRP1-dependent cross-reactivity observed in biochemical studies in cells in culture is buried in the background in living patients, whereas the 50-fold MDR1 Pgp-dependent transport signals translate well to patients, as has been reported by many clinical trials with 99mTc-sestamibi. Furthermore, cellular accumulation in the presence of specific inhibitors of MDR1 Pgp, such as LY335979, readily differentiate transport mediated by MDR1 Pgp versus MRP1 in vitro, and cells stably transfected with MRP2–MRP6 did not show any clear trends. These observations were consistent with the postulation that Ga-complex 1b will be a sensitive and reasonably selective probe for MDR1 Pgp transport activity in vivo.
MDR1 Pgp is expressed in capillary endothelial cells in the brain and forms a major component of the BBB (41). Therefore, mdr1a/1b(−/−) KO mice offer an interesting model to evaluate the utility of the reagent as a probe of Pgp transporter-mediated activity in vivo. Using the positron emissions of 68Ga, we readily detected greater net uptake in the brain of KO mice, consistent with the quantitative biodistribution data obtained with the 67Ga-complex. A drug-transporting protein, Pgp is also expressed along biliary canalicular surfaces of hepatocytes, poised to secrete substances into the bile. Biodistribution data in WT and KO mice demonstrated differences in accumulation of the radiotracer as expected for the presence or absence of drug-transporting Pgp in the liver.
PET agents exemplified by 11C-verapamil (57,58) and 18F-paclitaxel (18,19) have shown potential for noninvasive imaging of Pgp-mediated drug transport at the BBB. 11C-Verapamil demonstrated 10-fold enhanced activity in the brain of KO mice compared with that of WT mice. However, this PET agent did not show any significant differences of activity in other tissues, including the liver (58). Another PET agent, 18F-paclitaxel, also displayed pharmacokinetic profiles consistent with Pgp-mediated transport. The radiotracer showed enhanced accumulation in the brain of KO mice compared with that of WT mice with a differential uptake ratio (DUR = %ID/g × body weight) of 1,400 at 30 min after injection (18). However, the agent was similar to 11C-verapamil in the livers of KO and WT mice, displaying a lack of significant differential uptake, in contrast to 99mTc-sestamibi or 67/68Ga-complex 1b. This lack of differential uptake in the livers of KO mice compared with WT mice for 11C-verapamil and 18F-paclitaxel may be attributed to the existence of metabolites or cross-reactivity with other transporters in the tissue. In contrast, 67Ga-complex 1b showed a DUR of 1,700 at 60 min after injection in the brain and 205 in the liver. Furthermore, 67Ga-complex 1b showed 3- to 5-fold greater differences in brain AUC5–120 between KO and WT mice than 99mTc-sestamibi (59) and 99mTc-tetrofosmin (27), further documenting its potential superiority for imaging Pgp activity in vivo.
In addition, the liver showed twice the retained activity (AUC5–120) of 1b in KO mice compared with WT mice, whereas the heart, a Pgp-negative tissue, did not show any significant difference. Further analysis revealed that the heart-to-liver ratio in WT mice at 120 min increased to 2.2 compared with 0.4 in KO mice at the same time point. The observed lower heart-to-liver ratio in KO mice was consistent with a lack of cross-reacting non-Pgp–mediated clearance of 1b in KO mice. Furthermore, the heart-to-liver ratio of 2.2 for 67Ga-complex 1b reported herein compared favorably with 1.1 for 99mTc-sestamibi (59). Thus, as an additional consequence of the differential tissue expression of Pgp, the superior heart-to-liver ratio of 67Ga-complex 1b versus 99mTc-sestamibi at 2 h would be beneficial in myocardial perfusion imaging.
CONCLUSION
While exploring structure–activity relationships of N4O2 gallium(III) complexes, we have identified a nonmetabolized cationic, hydrophobic 67/68Ga-complex that is recognized by MDR1 Pgp as an efficient transport substrate. We are cautiously optimistic that this radiopharmaceutical may enable both SPECT and PET monitoring of Pgp activity in tissues in vivo. Issues related to radiation dosimetry, automation, or remote synthesis will require additional experimentation. The potential for a 68Ga generator-produced PET radiopharmaceutical for use within clinical environments, coupled with facile synthesis of the precursor ligand and a rapid radiolabeling procedure, enhances the promise of this approach for noninvasive assessment of Pgp at the BBB, in chemotherapy regimens, and during MDR gene therapy. Because of the mechanisms of cell localization and differential expression of Pgp in liver compared with heart, the agent also shows promise as a myocardial perfusion imaging agent.
Acknowledgments
The authors thank John Engelbach and Joon Young Kim for technical assistance with microPET imaging, the Research Resource for Isotope Research for 68Ga, and the Small Animal Imaging Resource (grant R24 CA83060) for small animal imaging support. This study was funded by National Institutes of Health Molecular Imaging Center grant P50 CA94056 and Department of Energy contract DE FG02 94ER61885.
Footnotes
Received Apr. 12, 2004; revision accepted Jul. 21, 2004.
For correspondence contact: David Piwnica-Worms, MD, PhD, Mallinckrodt Institute of Radiology, Washington University Medical School, 510 S. Kingshighway Blvd., Box 8225, St. Louis, MO 63110.
E-mail: piwnica-wormsd{at}mir.wustl.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}