


Abstract
Our objective was to develop an array of α7-selective nicotinic cholinergic receptor (nAChR)–based imaging agents for PET and SPECT. Methods: (2′R)-N-11C-Methyl-N-(phenylmethyl)-spiro[1-azabicyclo[2.2.2]octane-3,2′(3′H)-furo[2,3-b]pyridin]-5′-amine 1 was synthesized by reaction of the corresponding desmethyl precursor with 11C-CO2 and reduction. N-(R)-1-Aza-bicyclo[2.2.2]oct-3-yl-4-11C-methylsulfanyl-benzamide 2 was synthesized by reduction of the corresponding disulfide precursor and reaction with 11C-iodomethane. N-(R)-1-Aza-bicyclo[2.2.2]oct-3-yl-4-125I-iodo-benzamide 3 was synthesized by halogen exchange of the corresponding bromide. (2′R)-5′-(2-125I-iodo-3-furanyl)spiro[1-azabicyclo[2.2.2]octane]-3,2′(3′H)-furo[2,3-b]pyridine 4 was synthesized by the chloramine-T method. Kinetic biodistribution studies were done in male CD-1 mice by tail vein injection of 3.7 MBq (100 μCi) of the 11C-labeled radiotracer or 0.67 MBq (2 μCi) of the 125I-labeled radiotracer followed by brain dissection and tissue counting. Receptor blockade was determined by pretreatment of the mice with an excess of either unlabeled precursor or nicotine. Results: We synthesized 4 radiolabeled, moderate- to high-affinity, α7-nAChR–based ligands. The compounds were a series of quinuclidine derivatives with an inhibition constant (Ki) < 6 nmol/L (33 pmol/L for 4) for α7-nAChR and selectivities of α7/α4β2 subtypes of ≥14,000. All of the compounds were produced in adequate radiochemical yield and specific radioactivity (>74 GBq/μmol [2,000 Ci/mmol]). No site selectivity or receptor blockade was shown for 1 and 2 (0.91 ± 0.05 and 0.14 ± 0.03 %ID/g [percentage injected dose per gram] in the hippocampus [target tissue], respectively). Compound 3 showed low hippocampal uptake (0.25 ± 0.05 %ID/g) but prolonged retention within that structure. Pretreatment with nicotine decreased its uptake by up to 50% in the hippocampus. Similar reductions were also observed within the cerebellum (nontarget tissue). Compound 4 showed hippocampal uptake of 2.41 ± 0.03 %ID/g and target-to-nontarget uptake ratios of up to 2. Pretreatment of animals with unlabeled 4 resulted in a decrease of hippocampal uptake to 60% of its preblockade value without a corresponding decrease in cerebellar uptake. Conclusion: With further structural optimization, selective imaging of α7-nAChR may be possible.
The α7 nicotinic cholinergic receptor (α7-nAChR) is a cationic, ligand-gated calcium channel with 5 identical ligand-binding subunits (1). Along with the α4β2-nAChR subtype, the α7-nAChR represents the most abundant nAChR in the brain. Brejc et al. have recently elucidated the crystal structure of a related protein, the acetylcholine-binding protein (2). Presynaptically, α7-nAChRs modulate transmitter release, postsynaptically they are excitatory and generate depolarizing currents, and perisynaptically they provide a neuromodulatory function (1). Additional pharmacology includes influencing neurite outgrowth, activating second messenger systems, and facilitating memory—that is, long-term potentiation (3). The regulation of calcium-related events by α7-nAChRs underlies the ability for this nicotinic receptor subtype to modulate glutamatergic and nitrergic transmission (4). The α7-nAChR mediates increases in tyrosine hydroxylase expression on activation, conferring neuroprotection via the Src, Akt, Bcl-2/Bcl-x pathway (5) and facilitates amyloid deposition within neurons (6). In the central nervous system, α7-nAChRs are involved in sensory gating, memory, and neuronal plasticity. α7-nAChRs have also been implicated in a wide variety of pathologic states, including Alzheimer’s disease (7), schizophrenia (8), inflammation (9), and lung cancer (10). Notably, though the number of α4β2-nAChRs is decreased in Alzheimer’s disease, the number α7-nAChRs remains largely intact and therefore available for binding new therapeutic agents (7).
The 2 radiotracers used most commonly in vitro and ex vivo to characterize α7-nAChR are 125I-α-bungarotoxin (125I-α-BTX) and 3H-methyllycaconitine (3H-MLA), neither of which shows complete binding selectivity for α7-nAChR (11,12). 3H-MLA labels 21% more specific sites than does 125I-α-BTX, which displays slow binding kinetics due to its large size. These agents have been used to localize the α7-nAChR in the pons, hippocampus, and colliculi in the mouse brain (11). Specific ligands used to study α7-nAChRs range in size from that of the protein α-BTX down to AR-R17779, with a molecular weight of <200 g/mol. Several classes of compounds bind α7-nAChRs selectively, including oxystilbenes (13), anabaseines (14), and quinuclidines (15). Despite that diversity, structure–activity relationships pursued with the quinuclidine series indicated a low tolerance of the α7-nAChR for structural modification of putative ligands. Nevertheless, the pursuit of imaging agents based on the quinuclidines remains an attractive goal due to the synthetic accessibility of such ligands and the importance of developing a site-selective imaging agent for α7-nAChRs to complement those already developed for the α4β2-nAChR subtype (16).
Although a variety of compounds have been synthesized for α7-nAChRs (13,17), only 3 have been synthesized in radiolabeled form for imaging: 11C-AR-R255082 (18), 3-([2,4-dimethyl-5-123I-iodo]benzylidene)anabaseine (19), and 125I-MLA (20), with neither of the first 2 demonstrating any regionally selective binding in vivo. Navarro et al. have recently reported the synthesis of 125I-MLA and have shown encouraging in vivo biodistribution studies in rats, despite the apparently rapid clearance and poor brain penetration of this agent (20). Because α7-nAChRs are implicated in a wide variety of pathologic entities, and to further our ongoing program to image nAChRs, we have synthesized a series of potential site-selective α7-nAChR imaging agents based on the quinuclidine nucleus (15).
MATERIALS AND METHODS
Chemistry
N,N-Dimethylformamide was distilled under reduced pressure from BaO. 1H NMR spectra were obtained with a Varian 400 (400 MHz) instrument. Chemical shifts are reported in ppm (1H) relative to internal tetramethylsilane in CDCl3. High-resolution mass spectrometry was performed at the University of Minnesota Mass Spectrometry facility. Elemental analyses were determined by Quantitative Technologies Inc. Short-path column chromatography was performed using Merck 7729 (<230 mesh) silica gel. 11C-Iodomethane was prepared with the General Electric PETtrace MeI MicroLab using a PETtrace biomedical cyclotron. High-performance liquid chromatography (HPLC) equipment consisted of Rheodyne 7126 injectors, Waters 590 EF pumps, Waters 440 ultraviolet (UV) absorbance detector (254 nm), and a NaI(Tl) crystal (5.08 cm [2 in.]) scintillation detector. Hewlett-Packard 3390A integrators and a Rainin Dynamax system were used to record and analyze HPLC chromatograms. Semipreparative (10 × 250 mm) and analytic (4.6 × 250 mm) reversed-phase HPLC columns (Phenomenex Luna C-18, 10 mm) were used for purification and quality control of the radiotracers, respectively.
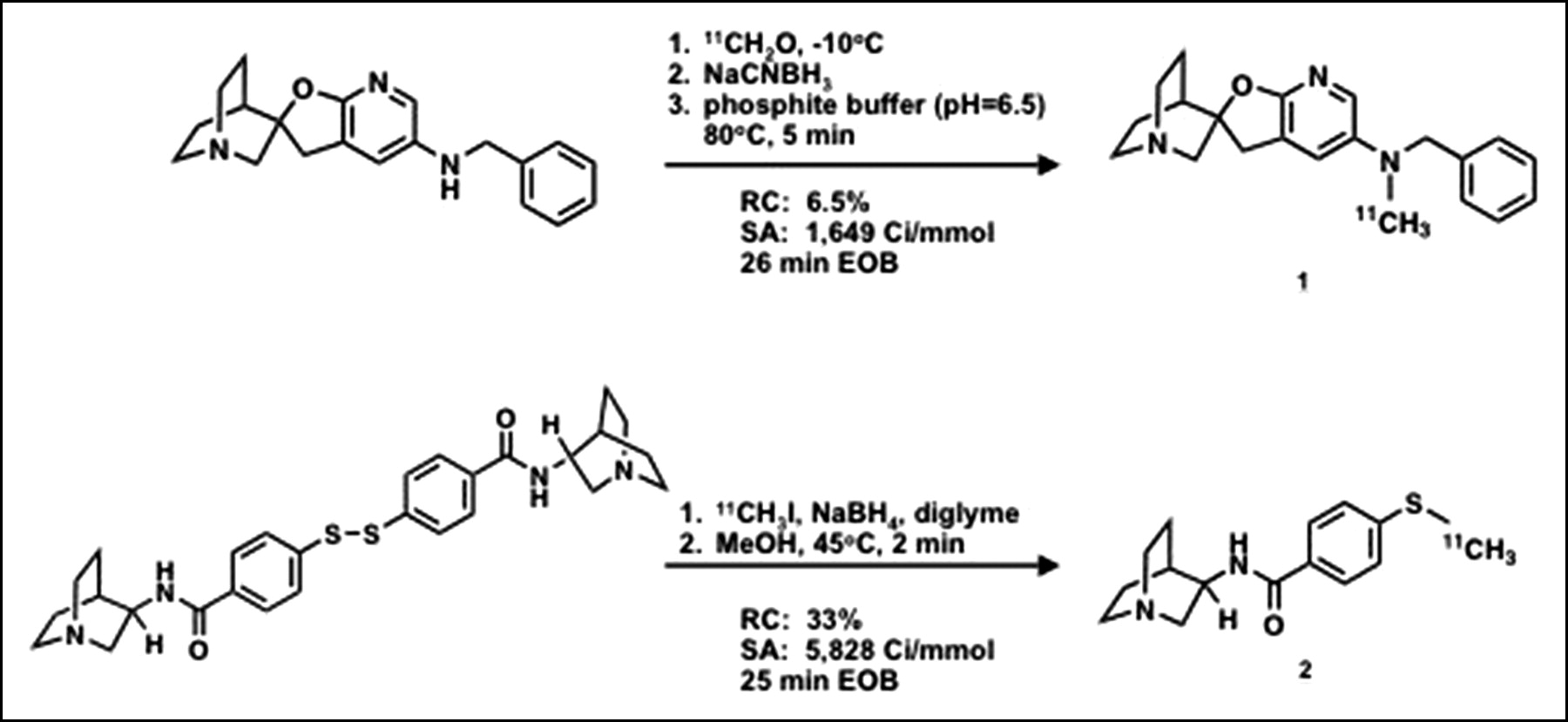
(2′R)-N-11C-Methyl-N-(Phenylmethyl)-Spiro[1-Azabicyclo[2.2.2]Octane-3,2′(3′H)-Furo[2,3-b]Pyridin]-5′-Amine 1 (Fig. 1).

11C-Labeled ligands: radiosyntheses. RC = radiochemical yield; SA = specific activity; EOB = end of bombardment.
The synthesis of the N-desmethyl precursor to 1, (2′R)-N-(phenylmethyl)-spiro[1-azabicyclo[2.2.2]octane-3,2′(3′H)-furo[2,3-b]pyridin]-5′-amine, has been described (21). 11C-Formaldehyde was generated in situ to synthesize 1 (22). 11C-CO2 was bubbled into a solution of lithium aluminum hydride in tetrahydrofuran (THF) (1.0 mol/L, 0.6 mL) at −10°C in a 15% sodium chloride ice bath, followed by the addition of 2 mol/L H2SO4 (0.6 mL). The ice bath was removed and 11C-CH2O was bubbled into a v-vial containing the desmethyl precursor (2.0 mg) in phosphite buffer, pH 6.5 (0.4 mL) (23). The reaction was heated at 80°C for 10 min before quenching with 500 μL of HPLC mobile phase consisting of 30:60 acetonitrile/water in 0.1 mol/L ammonium formate. The crude reaction was purified by reversed-phase HPLC using the mobile phase at 8 mL/min. The radioactive product (tR = 9.0 min), which was resolved from the precursor (tR = 5.3 min), was collected remotely. After concentration to dryness under reduced pressure and heat (80°C), the radiotracer was reconstituted in sterile 0.9% saline (7.0 mL) and passed through a 0.2-μm sterile filter (Acrodisc; Gelman) into a sterile, pyrogen-free multidose vial. Sterile NaHCO3 (3.0 mL, 8.4%) was added to give a final formulation of pH 7.4. Specific radioactivity was calculated by relating radioactivity to the mass associated with the UV absorbance peak of carrier.
N-(R)-1-Aza-Bicyclo[2.2.2]Oct-3-yl-4-11C-Methylsulfanyl-Benzamide 2 (Fig. 1).
The precursor to compound 2 was prepared by amide coupling of (R)-3-aminoquinuclidine (Sigma-Aldrich) with 4,4′-dithiobisbenzoic acid (Toronto Research Chemicals) using 1-hydroxybenzotriazole and O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate in dimethylformamide in the presence of diisopropylethylamine. 11C-Iodomethane, carried by a stream of nitrogen, was trapped in a dry ice/ethanol–cooled solution of the disulfide precursor (0.5–1.0 mg) in anhydrous MeOH (0.1 mL) and sodium borohydride in tetraglyme (3 mol/L, 0.1 mL). The reaction was heated at 45°C for 2 min before quenching with 200 μL of HPLC mobile phase consisting of 0.1% trifluoroacetic acid in 20:80 acetonitrile/water. The crude reaction was purified by reversed-phase HPLC using the mobile phase at 12 mL/min. The radioactive product (tR = 8.9 min), which was well separated from the precursor (tR = 5.5 min), was remotely collected. A single radioactive peak (tR = 2.3 min) corresponding to authentic 2 was observed. The radiotracer was formulated in the same manner as for 1. Specific radioactivities were calculated in a manner identical to that for 1.
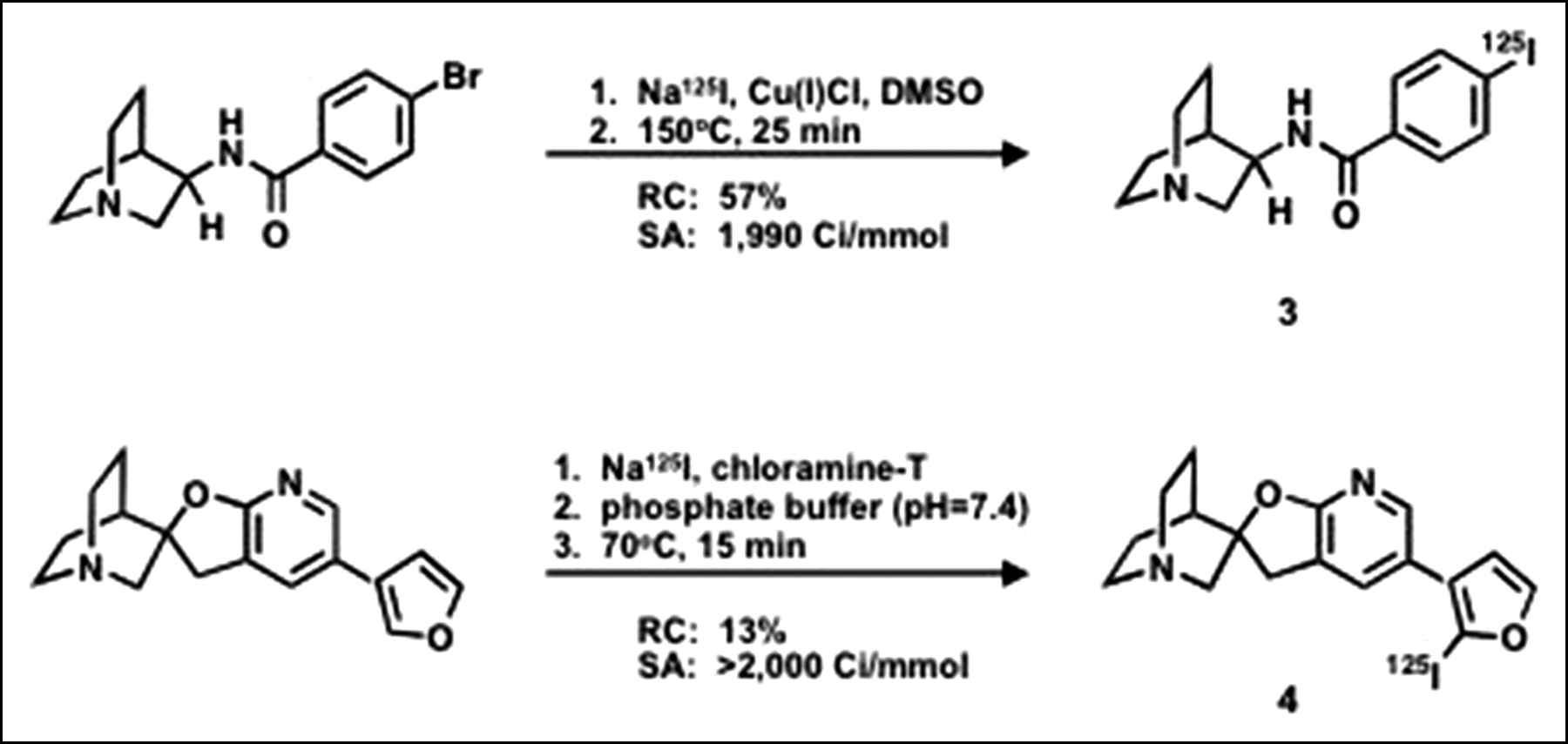
N-(R)-1-Aza-Bicyclo[2.2.2]Oct-3-yl-4-125I-Iodo-Benzamide 3 (Fig. 2).
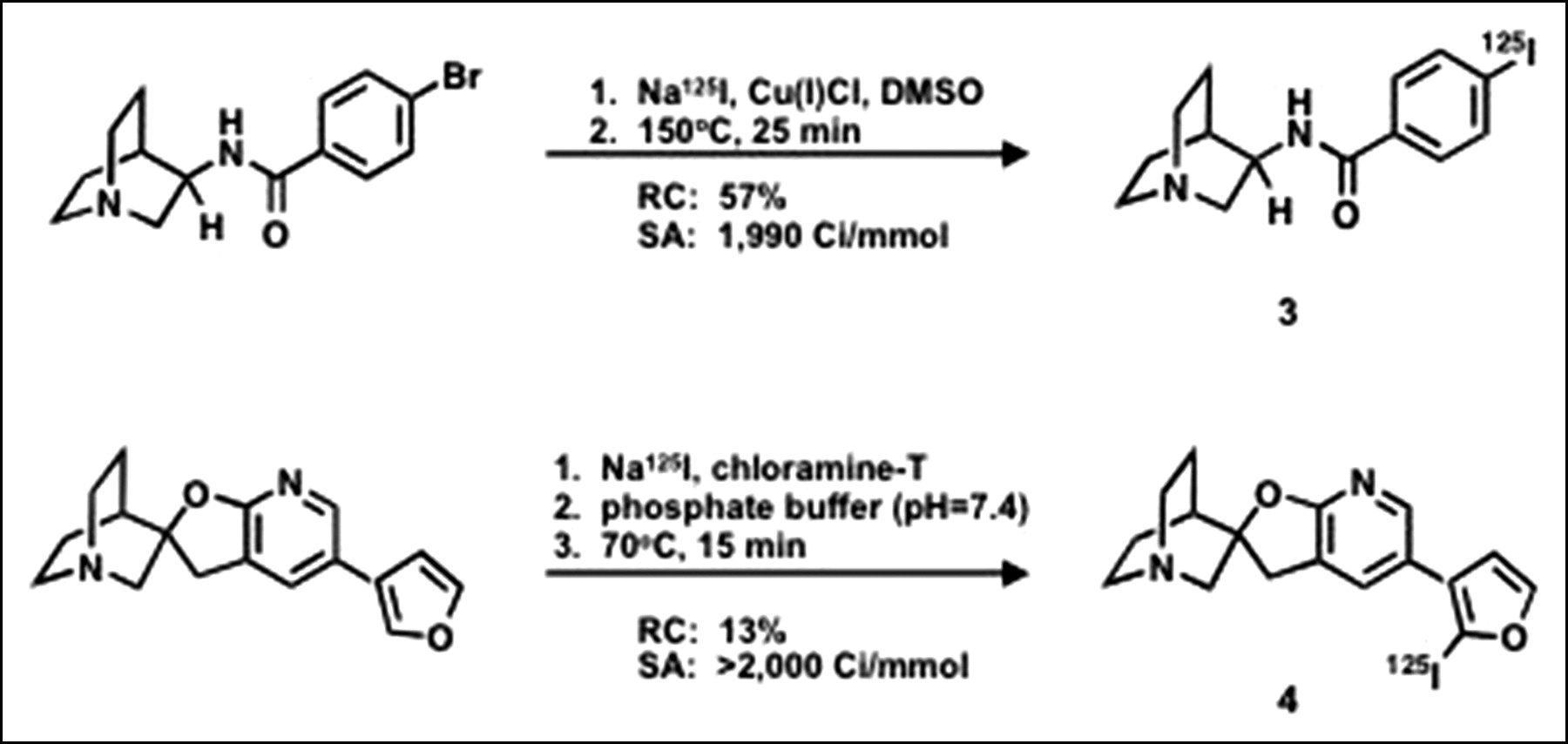
125I-Labeled ligands: radiosyntheses. DMSO = dimethyl sulfoxide; RC = radiochemical yield; SA = specific activity.
The free base of the brominated precursor, N-(R)-1-aza-bicyclo[2.2.2]oct-3-yl-4-bromobenzamide (24), to 3 was prepared by dissolving 1 mg in 0.4 mL of water, adding 0.1 mL of 0.1 mol/L NaOH, extracting with dichloromethane (0.3 mL), and evaporating the organic phase to dryness. The residue was taken up by methylsulfoxide (10 μL) and transferred to a 1-mL v-vial. To the vial were added 5 μL of 125I-NaI (11.7 MBq, 314 μCi) and 20 μL of a methylsulfoxide solution of Cu(I)Cl (10 mg/mL) (16,25). The sealed vial was placed in a 150°C sand bath and heated for 25 min. The reaction was cooled and subsequently quenched by addition of 0.4 mL mobile phase (25:75 acetonitrile/water in 0.1 mol/L ammonium formate). The crude reaction mixture was purified on a C-18 Luna column 10 × 250 mm at a flow rate of 6 mL/min. The desired radioactive peak (tR = 30 min) was well resolved from the bromo precursor (tR = 19 min) and the radiotracer was collected. The radioligand was concentrated by rotary evaporation at 45°C and formulated in saline (∼74 kBq [2 μCi] per 0.2 mL) for mouse biodistribution studies. A semipreparative C-18 Luna column using a mobile phase of 25:75 acetonitrile/water in 0.1 mol/L ammonium formate at 12 mL/min was used to estimate specific radioactivity and confirm radiochemical purity.
(2′R)-5′-(2-125I-Iodo-3-Furanyl)Spiro[1-Azabicyclo[2.2.2]Octane]-3,2′(3′H)-Furo[2,3-b]Pyridine 4 (Fig. 2).
To a 0.5-mL v-vial containing 25 μL of the spirofuropyridine precursor, (2′R)-5′-(2-3-furanyl)spiro[1-azabicyclo[2.2.2]octane]-3,2′(3′H)-furo[2,3-b]pyridine (26) (0.35 mg in 0.1 mL phosphate buffer), 125I-NaI (81 MBq, 2.2 mCi) and 25 μL of an aqueous solution of chloramine-T(2.3 mg/mL) were added. The sealed vial was placed in a 70°C sand bath and heated for 15 min. The reaction was cooled and subsequently quenched by addition of 50 μL of sodium metabisulfite (0.1 mol/L) and 100 μL of mobile phase (30:70 acetonitrile/water in 0.1 mol/L ammonium formate). The crude reaction mixture was purified on a C-18 Luna column 10 × 250 mm at a flow rate of 6 mL/min. The desired radioactive peak (tR = 8.9 min) was resolved from precursor (tR = 6.3 min) and the radiotracer was collected. The radioligand was concentrated by rotary evaporation at 45°C and formulated in saline (∼74 kBq [2 μCi] per 0.2 mL) for mouse biodistribution studies. The semipreparative C-18 Luna column using a mobile phase of 30:70 acetonitrile/water in 0.1 mol/L ammonium formate at 6 mL/min was used to estimate specific radioactivity and confirm radiochemical purity.
Synthesis of Unlabeled 4.
A solution of the spirofuropyridine precursor (0.9 mg, 3.2 μmol) in dichloromethane (70 μL) was added to mercuric acetate (1.5 mg, 3.3 μmol) in dichloromethane (70 μL). The mixture was stirred for 15 min. A solution of I2 (1 mg, 3.2 μmol) in dichloromethane was added dropwise and stirred for 30 min. The reaction mixture was diluted with chloroform (3 mL), filtered, washed with 5% sodium thiosulfate (5 mL), water (5 mL), brine (5 mL), and dried. The residue was purified by reversed-phase HPLC using a mobile phase consisting of 35:65 acetonitrile/water in 0.1 mol/L ammonium formate at 6 mL/min (1.2 mg; yield, 91.9%). 1H NMR (CDCl3, δ) 1.55–1.60 (m, 2H), 1.77–1.89 (m, 2H), 2.30 (t, J = 0.8 Hz, 2H), 2.90 (t, J = 7.2 Hz, 2H), 3.00–3.13 (m, 4H), 3.46 (t, J = 10.0 Hz, 2H), 6.52 (d, J = 2.0 Hz, 1H), 7.65 (d, J = 2.0 Hz, 1H), 7.66 (m, 1H), 8.18 (m, 1H). HRMS-CI: m/z calculated 409.0413; found 409.0439 (M+H)+.
Receptor-Binding Assays
The affinities of 1–4 for the rat α4β2- and α7-nAChRs were determined as described previously (15). Affinities for the rat 5-hydroxytryptamine type 3 (5-HT3) receptor (5-HT3R) were also determined as described previously (27). Binding reactions had a volume of 0.5 mL and were conducted in 96 deep-well plates. Reactions were incubated with shaking for the indicated time and temperature, and then the membranes were collected by filtration using GF/C filters pretreated for 2 h with 0.01% polyethyleneimine (PEI)/1% bovine serum albumin (BSA) solution for the α7-nAChR assay; GF/B filters pretreated for 2 h with 0.5% PEI were used for the α4β2-nAChR and 5-HT3R assays. The filters were washed 4 times with cold assay buffer and then counted for radioactivity in the presence of scintillation fluid. Assay conditions were as follows: α7-nAChR, 5 nmol/L 125I-α-BTX, rat hippocampal membranes, 0.1 mg/mL BSA buffer (pH 7.4), and incubated for 2 h at 20°C; α4β2-nAChR, 3 nmol/L 3H-nicotine, rat cortical membranes, buffer, and incubated for 1 h at 4°C; 5-HT3R assay, 0.5 nmol/L 3H-zacopride, rat small bowel muscularis membranes, 150 mmol/L NaCl, 50 mmol/L Tris·HCl, pH 7.4, and incubated for 1 h at 37°C. Note that for compound 4, the 5-HT3R assay used ha7-HEK membranes and 0.3 nmol/L 3H-GR65630 (28), and samples were incubated for 1 h at 20°C.
Fifty percent inhibitory concentration (IC50) values were determined from 5–7 drug concentrations (triplicates). Inhibition constant (Ki) values were determined using the Cheng–Prusoff equation: Ki = [IC50]/(2 + ([ligand]/Kd)n)1/n − 1, where Kd is the dissociation constant, n = 2 for the α7-nAChR–binding assay, and n = 1 for α4β2-nAChR and 5-HT3R assays (29). The SEM was obtained for values determined >3 times. Reference compounds used in these assays include nicotine (α4β2-nAChR), epibatidine (α4β2-nAChR), AR-R17779 (α7-nAChR), tropisetron (5-HT3R and α7-nAChR), ondansetron (5-HT3R), and LY-278,584 (5-HT3R).
Physical Properties of 1–4
ACD log P was calculated using a log P prediction method as implemented in ACD UNIX Batch V7.0 (Advanced Chemistry Development, Inc.). It is also based on a fragment contribution method. More information can be obtained at: http://www.acdlabs.com/products/phys_chem_lab/logp/
Animal Studies: Rodent In Vivo Biodistribution Studies of 1–4
All animal studies were approved by the Animal Care and Use Committee of the Johns Hopkins University. Male CD-1 mice weighing between 20 and 25 g received an injection of 3.7 MBq (100 μCi) for 1 and 2 and 0.67 MBq (2 μCi) for 3 and 4 through the tail vein. The corresponding amounts administered were 0.2–0.8 μg/kg for 1 and 2 and 0.01–0.02 μg/kg for 3 and 4. For kinetic studies of 1–4, the mice were killed by cervical dislocation at 5, 15, 30, and 60 min after intravenous injection of radiotracer in 200 μL saline vehicle. The brains were removed and placed on ice, and the cerebellum, hippocampus, striatum, cortex, and thalamus were harvested for all compounds. For 1 and 4, the superior colliculus was also harvested. For 2 and 3, the hypothalamus was harvested, and the brain stem was obtained for 3 and 4. Olfactory bulb was obtained only in the case of 3. A 45-min time point was obtained for 2 and a time point was obtained at 120 min for 3 and 4. The tissue samples were weighed and their radioactivity was determined in an automated γ-counter (1282 Compugamma CS; Pharmacia/LKB Nuclear Inc.). The radioactivity concentration in aliquots of the injected tracer was determined along with the samples and served as standards for the calculation of the percentage injected dose per gram of tissue (%ID/g). To assess binding specificity, subgroups of mice were treated with low-specific-activity (LSA) radiolabeled material (for 1), in which unlabeled 1 was added to the radiolabeled injectate to provide a coinjectate of radiolabeled material and 1 mg/kg of unlabeled 1, 10 mg/kg nicotine, subcutaneously (for 3), or increasing doses of unlabeled 4 (0.01–10 mg/kg, intraperitoneally) 5 min before injection of radiotracer. Mice were killed at 30 and 60 min after blockade for 3 and 4, respectively.
Statistics
ANOVA, which was used in the biodistribution studies, was performed with StatView SE + Graphics software, version 5.0 (SAS Institute). For the paired Student t test, P < 0.05 was considered statistically significant.
RESULTS
Radiochemical Syntheses
After reaction with 11C-CH2O, generated in situ from 11C-CO2, compound 1 was produced in 6.5% radiochemical yield at 26 min after end of synthesis (EOS) (Fig. 1). A specific radioactivity of 61 GBq/μmol (1,649 Ci/mmol) was obtained. The disulfide bond of the precursor to 2 was reduced and the resulting thiol reacted with 11C-iodomethane to give the radiomethylated adduct in 33% yield with specific radioactivities that ranged from 152 to 216 GBq/μmol (4,095–5,828 Ci/mmol) with an average (n = 2) of 184 GBq/μmol (4,962 Ci/mmol) calculated at the EOS (Fig. 1).
Compound 3 was synthesized from the quinuclidyl bromobenzamide precursor by a copper-assisted iodo-debromination in 57% yield at a specific radioactivity of 74 GBq/μmol (1,990 Ci/mmol) (Fig. 2). Compound 4 was made by treatment with 125I-NaI according to the chloramine-T method in 13% yield at a specific activity of >74 GBq/μmol (>2,000 Ci/mmol) (Fig. 2). An unlabeled analog of 4 was synthesized and characterized to provide a standard for HPLC.
Receptor-Binding Affinities and Selectivities
The relative affinities (Ki values) of the nonradioactive analogs of 1–4 ranged from 0.26 to 18 nmol/L (Table 1). There was a wide margin in selectivity compared with the α4β2 receptor subtype. Also included are the relative binding affinities to the 5-HT3R, which has high sequence homology to the α7-nAChR (17,30). Binding selectivity to α4β2-nAChR was not performed for 4. By the time 4 was synthesized, the α4β2-nAChR assay was no longer performed routinely for this series because of the demonstrated reliable selectivity to α7-nAChR over α4β2-nAChR (Table 1).
Binding Affinities and Selectivities
Physical Properties of 1–4
The physical properties of 1–4 are depicted in Table 2. The log P values and molecular weights indicate that those compounds should gain ready access to the brain.
Physical Properties of Potential Ligands
Rodent Brain Biodistribution Studies
We observed no α7-nAChR–selective, regional brain uptake at 5, 15, 30, and 60 min for 1 (Table 3). Also shown are uptake values for LSA determinations at 30 and 60 min. No regional selectivity was demonstrated, nor was there evidence of receptor blockade in the LSA studies. As with the other radiotracers in this study (except for 2), compound 1 obtained limited but measurable uptake within the brain. We also observed limited uptake of 2 within the brain at all time points, with no regional selectivity (Table 4). Although 3 does not attain high uptake values within the brain, at values < 0.4 %ID/g, retention within hippocampus, a target tissue, was demonstrated (Table 5; Fig. 3), including significantly higher levels of hippocampal and cortical uptake at 120 min than seen for cerebellum (P = 0.012 and 0.005, respectively). That observation was sufficiently encouraging to warrant a blocking study that failed to demonstrate regionally selective blockade, although significantly decreased radiotracer uptake was demonstrated in the cerebellum (P = 0.005), hippocampus (P < 0.0001), striatum (P = 0.002), and cortex (P = 0.0002) (Fig. 4). Compound 4 gained ready access to the brain with modest but higher uptake in target tissue (hippocampus and superior colliculus) than within other brain substructures (Table 6; Fig. 5). Specifically, the hippocampus, superior colliculus, and cortex demonstrated higher uptake than the cerebellum at 60 min (P = 0.013, 0.021, and 0.014, respectively). The receptor blockade study indicates a significant blockade in the target tissue (hippocampus, P = 0.002; superior colliculus, P = 0.017) as well as in the cortex (P = 0.010), which also contains α7-nAChRs (Fig. 6).

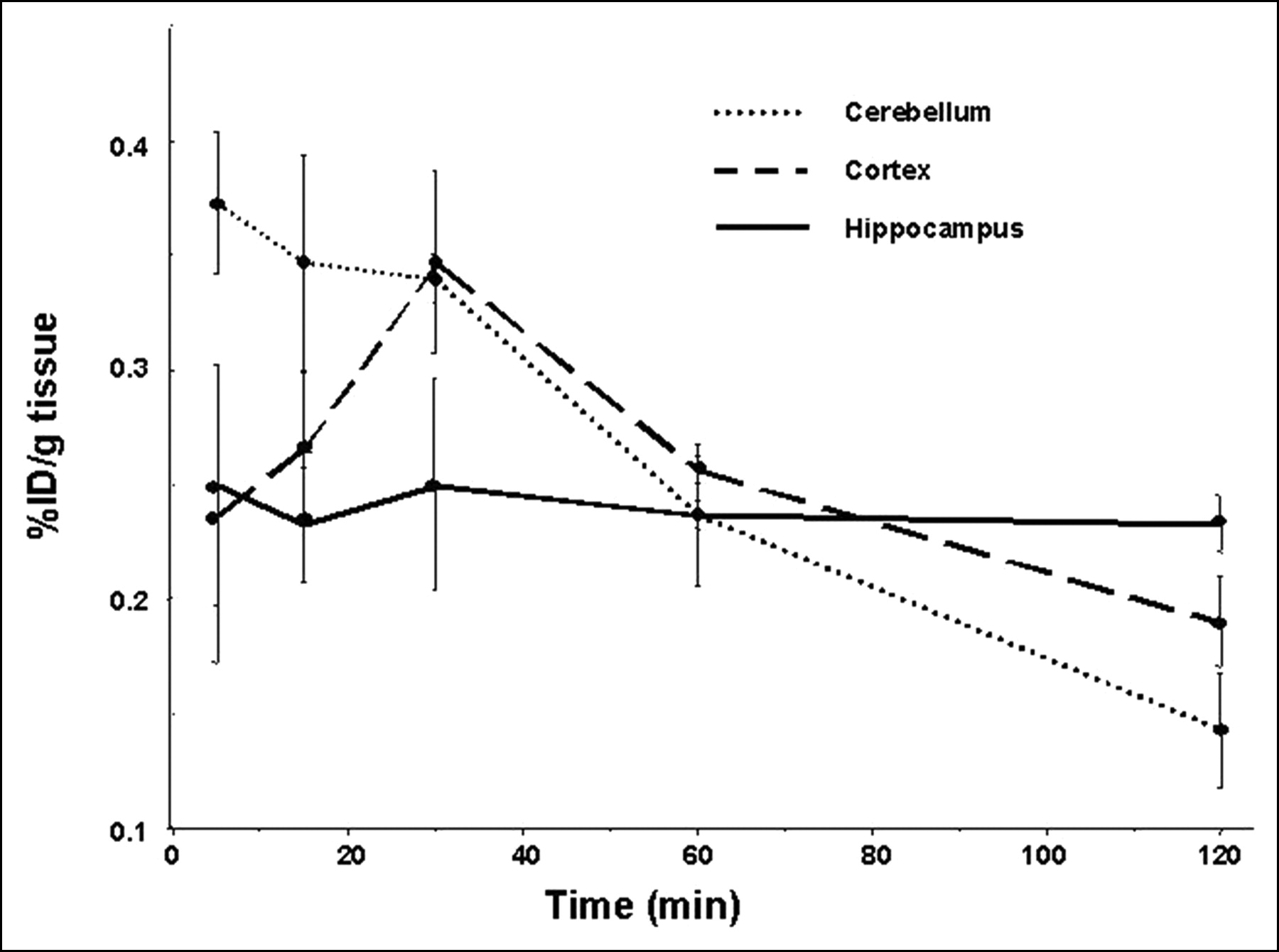
Brain kinetics of 3. Time course of uptake of 3 is presented in cerebellum (low α7-nAChR content), cortex (intermediate α7-nAChR content), and hippocampus (target tissue). Although up to 80 min after injection there is less uptake demonstrated in hippocampus than in cerebellum or cortex, hippocampal uptake remains constant and persists over time.

Binding specificity of 3. A significant decrease in uptake of 3 was demonstrated in cerebellum, hippocampus, striatum, and cortex on pretreatment with 10 mg/kg nicotine to block α7-nAChR. Because blockade was demonstrated within each region, including cerebellum, a nontarget region, we cannot conclude that the degree of blockade demonstrated can all be attributed to specific binding.
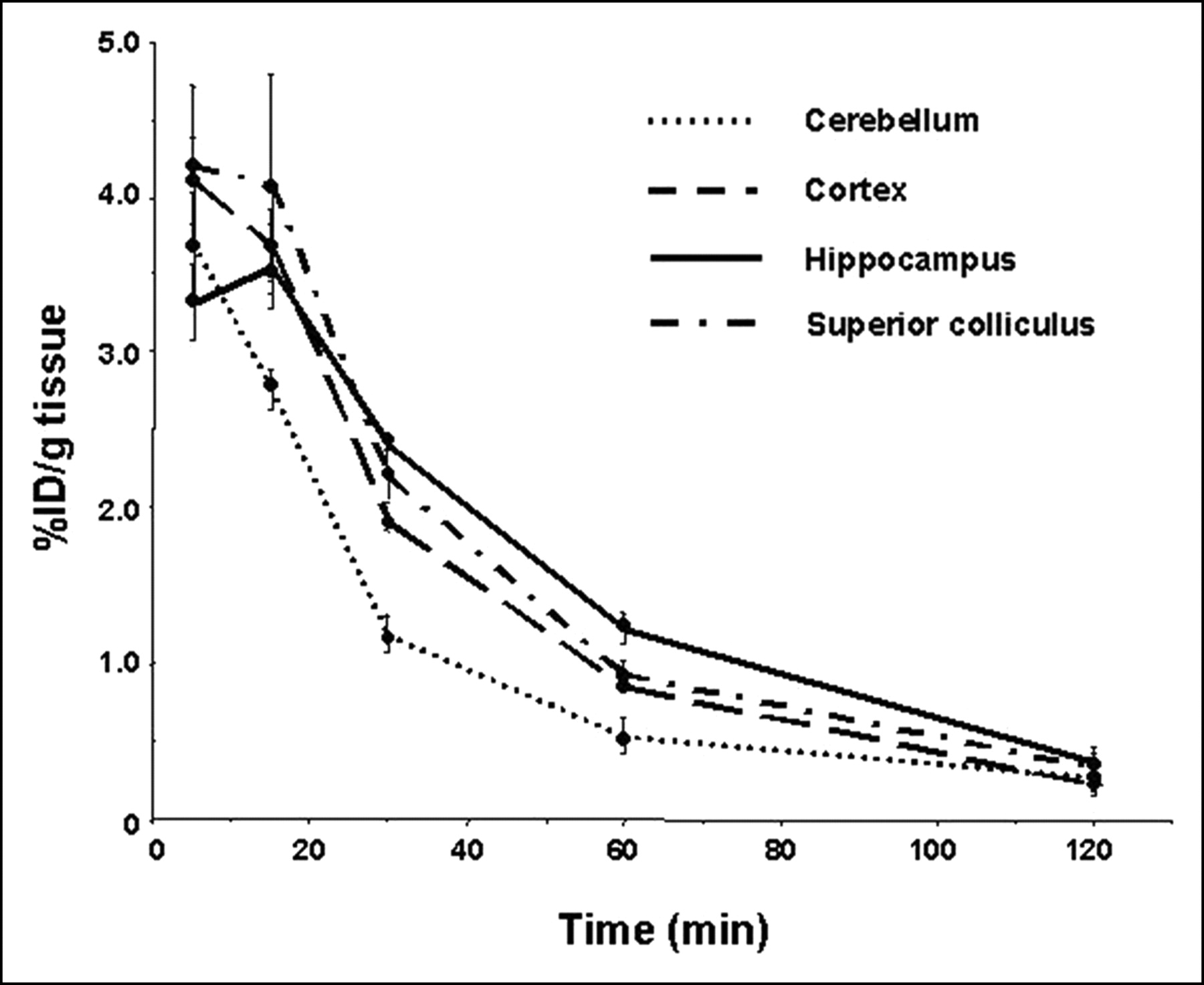
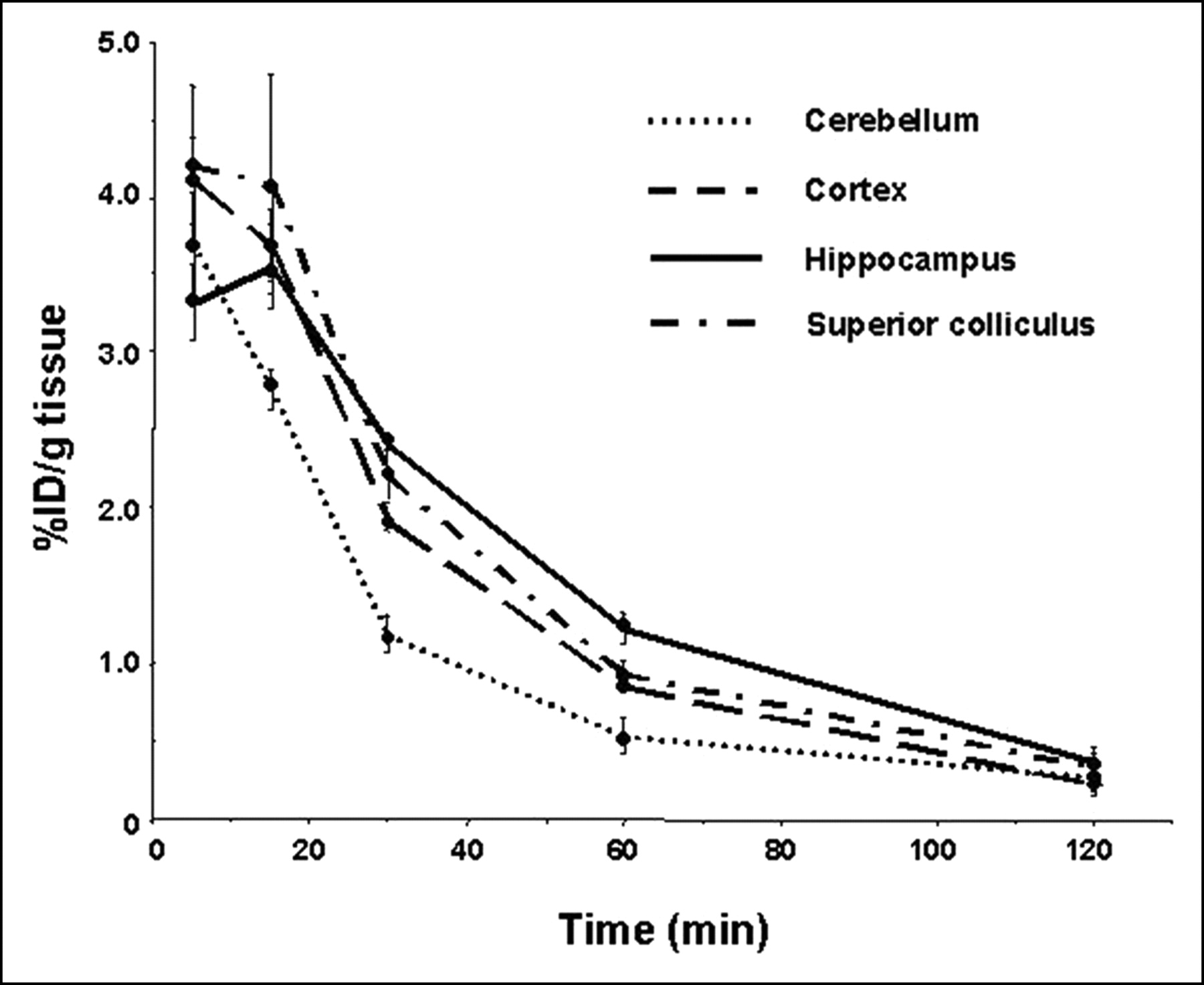
Brain kinetics of 4. Time course of uptake of 4 is presented in cerebellum, cortex, hippocampus, and superior colliculus (another target tissue). Note that higher uptake of 4 is demonstrated in target tissues over the entire time course of this study.

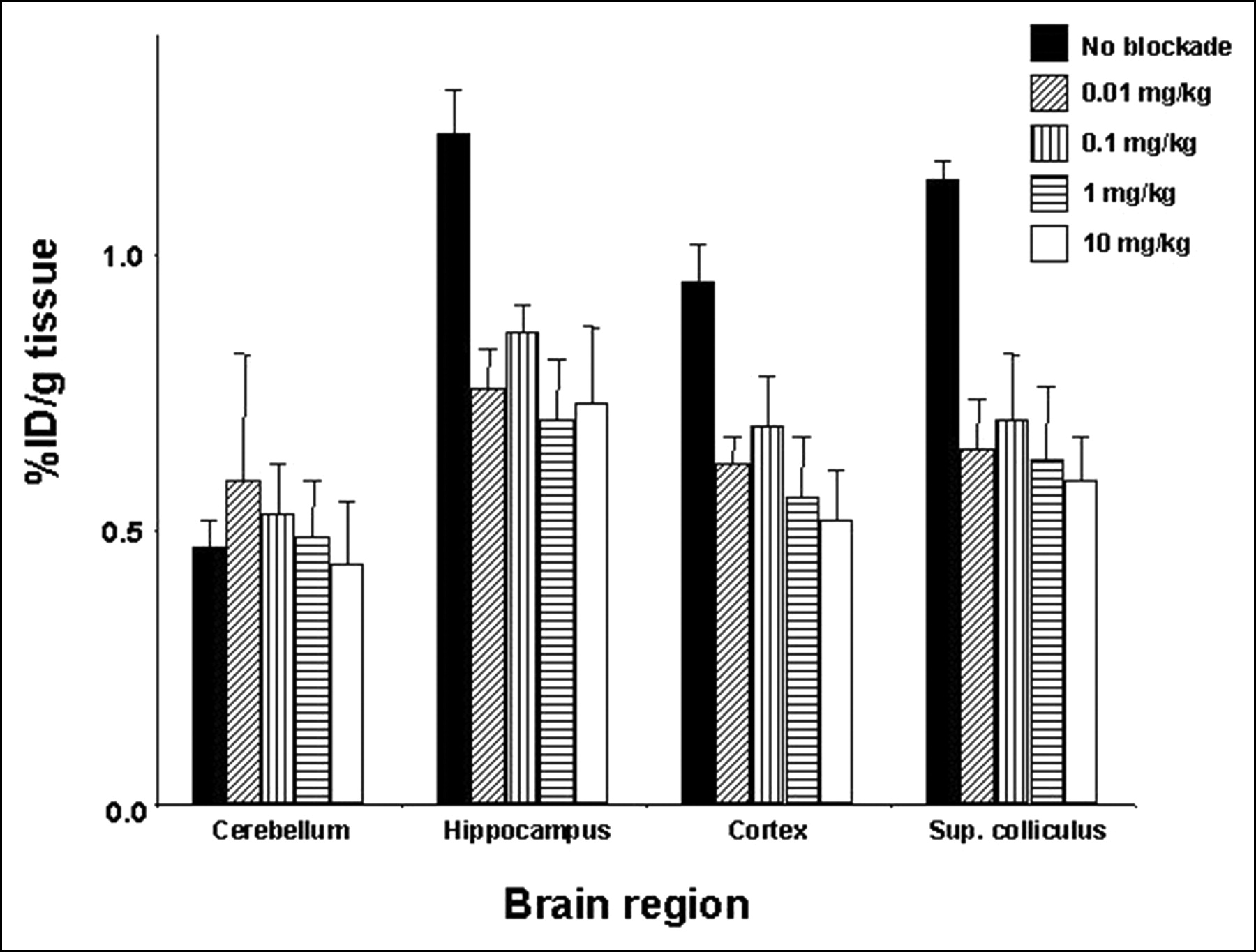
Binding specificity of 4. A significant decrease in uptake of 4 was obtained in all regions tested except for cerebellum, which has low α7-nAChR content, on pretreatment with increasing concentrations of unlabeled 4. These findings suggest an element of specific binding of 4 to α7-nAChR. Sup. colliculus = superior colliculus.
Brain Distribution of Compound 1
Brain Distribution of Compound 2
Brain Distribution of Compound 3
Brain Distribution of Compound 4
DISCUSSION
The goal of this work was to synthesize γ-emitting ligands that demonstrate selective binding to the α7-nAChR that could be further developed into imaging agents for PET and SPECT. Target sites for the α7-nAChR within the human brain include the hippocampus (CA1 region), entorhinal cortex, and superior colliculus (31). α7-nAChRs also tend to be evenly distributed throughout the cortex. The brain distributions of human and rodent α7-nAChRs correlate (11). Although most studies of α7-nAChR distribution in brain are qualitative, one study that used 125I-MLA showed a maximum number of binding sites of 68 ± 3 fmol/mg protein in rat cerebral cortex and another, using 3H-MLA, demonstrated 59 ± 4 fmol/mg in mouse hippocampus (11,20). Those concentrations are comparable to or higher than values for other receptors successfully imaged with PET and SPECT in human subjects and suggest that ligands with low nanomolar affinities, comparable to compounds 1, 2, and 4 (Table 1), should be successful for imaging of α7-nAChR in vivo (32,33).
Although the binding affinities of the compounds tested, except perhaps for compound 3, are at least moderate (Table 1), access to the brain remained low. On the basis of their physical properties, those compounds should readily cross membranes (Table 2). Rapid metabolism or active pumping out of the brain could also account for the poor brain penetration. Compounds 1 and 2 showed such a lack of regional selectivity that only a preliminary dose-saturation (receptor selectivity) study was performed in the case of 1—that is, by performing uptake with LSA radiotracer (Tables 3 and 4). Those compounds are not suitable for further development as radiopharmaceuticals for PET.
For 3, which demonstrated intermediate brain access, prolonged retention was identified within the hippocampus, suggesting an element of specific binding (Table 5; Fig. 3). Because brain uptake was actually less in the hippocampus than within nontarget sites at 30 and 60 min, and because no specific blockade could be demonstrated, 3 was also deemed not suitable for further development as an imaging agent. However, we believe that at least an element of the blockade demonstrated with 10 mg/kg of pretreatment with nicotine was due to receptor-specific blockade because nicotine generally causes an increase in regional cerebral blood flow (rCBF) (34). Usually such receptor blockade engenders a decrease in rCBF so that decreased delivery (or increased washout) may be the reason for decreased radiotracer uptake in those cases, rather than specific receptor blockade. Nicotine, however, may have caused increased washout of the radiotracer in this case.
Compound 4 showed the most promise of this series by demonstrating a reasonable degree of brain uptake (approximately 2.5% and 1.5% at 30 and 60 min, respectively), higher uptake in the hippocampus and superior colliculus than in nontarget sites, and definite site-selective blockade (Fig. 6). Unfortunately, it is not entirely clear if it was the α7-nAChR or 5HT3R sites that were blocked because of the low binding selectivity of 4 for α7-nAChR over 5HT3R (Table 1). Because the uptake values for 4 parallel more closely the known brain distribution of α7-nAChR—that is, hippocampus ≥ superior colliculus > cortex > cerebellum (Fig. 5) rather than that for 5HT3R (cortex > hippocampus > superior colliculus), we are probably observing an element of α7-nAChR–selective binding (20,35). Further in vivo studies—that is, blocking with nicotine or 5HT3R ligands rather than autoblockade as was undertaken here—might determine to which receptor 4 was binding; however, the poor in vitro selectivity and modest regional preference for target sites did not warrant further development of this compound for imaging.
Notably, the compounds with highest affinity (1 and 4) also demonstrate the lowest binding selectivity relative to 5-HT3R. That is due to the high (30%) sequence homology and pharmacologic cross-reactivity between these 2 receptor systems, which are both members of the superfamily of ligand-gated ion channels (17,36,37). Nevertheless, differences have been identified within the ligand-recognition regions for 5-HT3R and α7-nAChR, and there are distinct pharmacologic properties of these 2 receptor subtypes (17). Dramatic changes in functional activity at these 2 receptor subtypes have been demonstrated on only minor structural changes of the ligands, underscoring the need for a better understanding of this cross-reactivity for the development of site-selective diagnostic and therapeutic agents (38). Fortunately, for the purposes of imaging, ondansetron, a selective 5-HT3R agonist that is used clinically for the treatment of nausea due to cancer chemotherapy, can in principle be administered before an α7-nAChR–based imaging agent to effect selective imaging at α7-nAChRs (27,39).
The synthesis of conformationally rigid analogs of flexible ligands such as nicotine represents a classical strategy to obtain receptor subtype selectivity (40). The spirofuropyridine system, exemplified by 1 and 4, provides a rigid analog of acetylcholine and provides the highest affinity ligands in this series. Further functionalization of the pyridine moiety seems in order to provide improved ligands for α7-nAChR. A combination of better understanding of the overlapping pharmacology of α7-nAChR and 5-HT3R, greater knowledge of the binding site of the α7-nAChR—which may be more readily forthcoming due to recent publication of the crystal structure of the acetylcholine-binding protein (2)—and further structural modification of analogs related to 4 may provide suitable imaging agents for α7-nAChR.
CONCLUSION
Radiolabeled α7-nAChR–selective ligands based on the quinuclidine nucleus can be synthesized in adequate radiochemical yields and with high specific radioactivities. Target tissue selectivities of compounds of that series are modest, but present. Future structure–activity studies will focus on the spirofuropyridine system (compound 4) with further input from recently available data from the x-ray crystal structure of the acetylcholine-binding protein.
Acknowledgments
We thank Paige Rauseo for technical assistance and National Institutes of Health grants CA92871 and DA13199 for financial support.
Footnotes
Received Jul. 5, 2004; revision accepted Sep. 24, 2004.
For correspondence or reprints contact: Martin G. Pomper, MD, PhD, Department of Radiology, Johns Hopkins University, 600 N. Wolfe St., Phipps B-100, Baltimore, MD 21287-2182.
E-mail: mpomper{at}jhmi.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}