


Abstract
The presence of a high density of somatostatin receptors (SSRs) on human tumors forms the basis for the successful visualization of primary tumors and their metastases using radiolabeled somatostatin analogs. In recent years somatostatin analogs, coupled to β-emitting radioisotopes, have been successfully applied in the treatment of patients with metastatic SSR-positive neuroendocrine tumors. This concept of targeting SSR-expressing tumors using peptide receptor radionuclide therapy may also apply to the use of somatostatin analogs coupled to chemotherapeutic compounds. Evidence for the effectiveness of such cytotoxic somatostatin analogs as antitumor agents has been provided in a significant number of studies in experimental tumor models. In addition to cytotoxic somatostatin analogs, somatostatin analogs coupled to peptides containing arginine, glycine, and aspartate and coupled to paclitaxel have been synthesized. Here we discuss the development of the different cytotoxic somatostatin analogs and their antitumor effects in vitro and in vivo in experimental models.
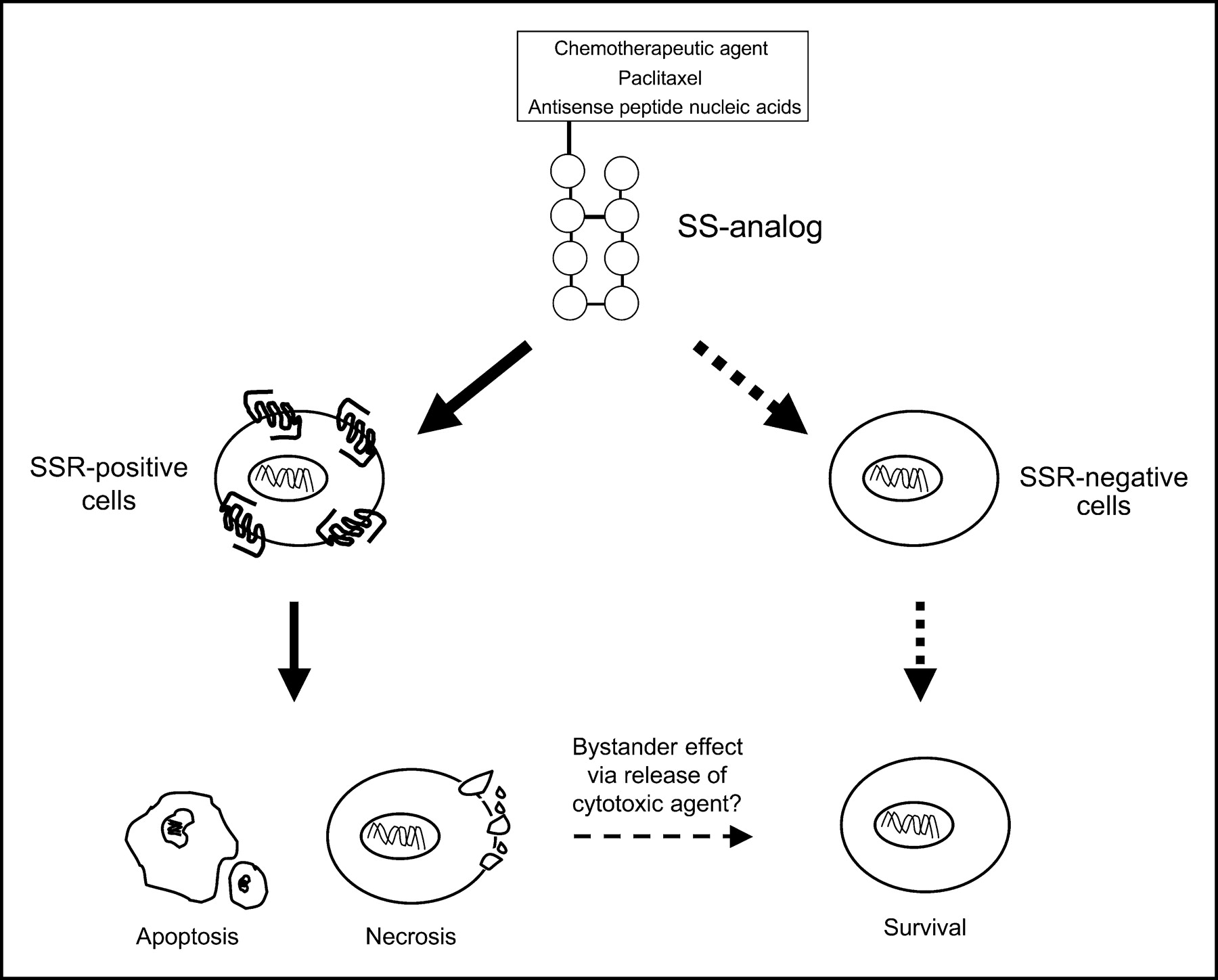
Toxicity of chemotherapeutic drugs is one of the main restrictions in the adjuvant treatment of patients with advanced or metastatic cancers. The concept of reducing toxicity via targeted delivery of cytotoxic agents was first proposed more than a decade ago. Schally and Nagy (1,2) developed a series of targeted antitumor agents by linking various cytotoxic radicals to analogs of luteinizing hormone-releasing hormone (LHRH), somatostatin, and bombesin. Targeted delivery of chemotherapeutic compounds coupled to small peptide molecules requires the presence of specific receptors on the target cancer cells. After binding of the cytotoxic peptide to its receptor, the hybrid molecule must be internalized by the tumor cells, after which the cytotoxic agent may be uncoupled from its linker and thus produce lethal action (Fig. 1). The aim of targeted chemotherapy in oncology is to improve effectiveness via the selective delivery of cytotoxic compounds to primary tumors and their metastases and to reduce peripheral toxicity. In addition to chemotherapeutic compounds, other molecules capable of modulating (tumor) cell growth can be coupled to somatostatin analogs. One group of small molecules includes Arg-Gly-Asp (RGD) molecules, which bind with high affinity to the integrin αvβ3 and have been shown to be inhibitors of cell adhesion, migration, growth, and differentiation. Again, via targeted delivery of such RGD somatostatin molecules to tumor cells, as well as to neovascular endothelial cells known to express a high density of αvβ3 receptors, these molecules may be potentially interesting antitumor agents. In this paper, the development and antitumor action of different cytotoxic somatostatin analogs as well as the development and preliminary characterization of radiolabeled RDG-coupled somatostatin analogs is reviewed.

Principle of SSR-targeted cytotoxic therapy.
SOMATOSTATIN RECEPTOR SUBTYPE EXPRESSION IN TUMOR TISSUES
Neuroendocrine tumors, which often originate from somatostatin-target tissues, frequently express a high density of somatostatin receptors (SSRs). Human tumors that express SSRs include pituitary adenomas, islet cell tumors, carcinoids, paragangliomas, pheochromocytomas, small cell lung cancers (SCLC), and medullary thyroid carcinomas, as well as malignant lymphomas and breast, prostate, and ovarian cancers (3). The SSR subtype expression in different types of human cancers has been demonstrated at the mRNA level using in situ hybridization, RNAse protection assays, the reverse transcriptase polymerase chain reaction (RT-PCR), and autoradiography, as well as by immunohistochemistry (3). The majority of human SSR-positive tumors simultaneously express multiple SSR subtypes, although there is a considerable variation in SSR subtype expression between different tumor types and among tumors of the same type. SSR autoradiographic studies showed the absence of binding of the SSR subtype 2 (SST2)-preferring analog 125I-Tyr3-octreotide in a small subgroup of human insulinomas, carcinoids, pituitary adenomas, and meningiomas; in 50% of medullary thyroid carcinomas (MTC); in prostate cancer; and in all SSR-positive ovarian cancers, whereas binding sites for radioiodinated somatostatins are present in the same tumors (3). In such cases, targeting tumors with SST2-preferring cytotoxic somatostatin analogs may not be successful. In addition to tumor cells, blood vessels, immune cells, and stromal cells, which are present in or surround human tumors, may express SSR subtypes (3). When targeted therapy using somatostatin analogs coupled to chemotherapeutic agents or RGD is considered, it is important to realize that some tumors may have a heterogeneous distribution of SSR. In particular, in more than 50% of breast cancer specimens SSR expression shows a nonhomogenous distribution, with regions of high density of SSR next to regions lacking the receptor (3). Knowledge of the SSR subtype expression pattern, as well as nonhomogeneity in the expression of SSR within tumors, is very important for the development of the concept of SSR-targeted chemotherapy. SSR subtypes differ in their ability to internalize receptor-bound ligand, a crucial step to direct a somatostatin analog linked to a cytotoxic compound or RGD into the internal milieu of the tumor cell, where it can exert its cytotoxic action (4).
INTERNALIZATION OF SOMATOSTATIN RECEPTOR SUBTYPES
Since the cloning of the 5 SSR subtypes, the involvement of the individual human SSR subtypes in the process of receptor-mediated internalization of SS has been extensively investigated. As indicated, the ability of SSR to internalize the ligand-receptor complex after ligand binding forms the basis for the concept of targeted chemotherapy using cytotoxic somatostatin analogs, as well as for the use of radiolabeled RGD–somatostatin analogs to selectively inhibit tumor growth. In general, the mechanism and route of internalization of SSR–agonist complexes follow those described for many other G-protein–coupled receptors and involve aggregation of the hormone receptor complex in specialized areas of the membrane, followed by internalization of the hormone–receptor complex via clathrin-coated as well as uncoated pits (5–9). After internalization and pit formation, fusion of these vesicles with lysosomes occurs, resulting in hormone degradation or receptor recycling to the cell surface (7,10,11). At this stage, the cytotoxic molecules are released and may exert their lethal action.
SSR subtypes differentially internalize somatostatin and somatostatin analogs (12). In Chinese hamster ovary (CHO)-K1 cells stably expressing 1 of the 5 human SSR subtypes, SST2, SST3, SST4, and SST5 receptors displayed rapid (within minutes) agonist-dependent internalization of 125I-LTT somatostatin-28 ligand in a time- and temperature-dependent manner (13). Maximum internalization of the radioligand occurred within 60 min. SST3- and SST5-expressing cells displayed the highest degree of internalization (78% and 66%, respectively), followed by SST4 (29%) and SST2 (20%). In contrast, the SST1 subtype displayed only a very low amount (4%) of internalization. Another study using COS-7 cells transfected with the human SST1 or SST2A receptor subtypes recently confirmed the low internalization rate of the SST1 subtype (14). However, in this study, up to 75% of specifically bound fluorescent ligand was recovered inside SST2A-expressing cells within 60 min after agonist exposure, where it clustered into small endosome-like particles (14). The capacity of internalization of somatostatin via the SST5 receptor was intermediate between SST1 and SST2A receptors (15).
A predominant importance of the SST2A receptor, which is widely expressed in many human tumors, in determining the uptake of 111In-diethylenetriaminepentaacetic acid0 (DTPA0)-octreotide in vivo is further underlined by the observation that uptake of 111In-DTPA0-octreotide in SSR-positive organs, such as the pituitary gland, adrenals, and thymus, is reduced by more than 80% in SST2A receptor knockout mice, compared with the values of uptake in wild-type mice carrying SST2A receptors (16). Moreover, a significant number of in vivo studies in experimental tumor models have shown that uptake of 111In-DTPA0-octreotide is blocked by the simultaneous administration of excess unlabeled octreotide (16–19). Finally, relative uptake values of radioactivity in patients with carcinoid tumor or neuroblastoma correlate with tumoral SST2 expression level (4). The ability of SSR subtypes to undergo agonist-induced internalization is an important characteristic of these receptors for transporting radiolabeled and cytotoxic somatostatin analogs into the cell.
DEVELOPMENT AND IN VITRO CHARACTERIZATION OF CYTOTOXIC SOMATOSTATIN ANALOGS
The first cytotoxic somatostatin analog described was methotrexate linked to the N-terminal of the octapeptide somatostatin analog RC-121 (d-Phe-Cys-Tyr-d-Trp-Lys-Val-Cys-Thr-NH2). This compound was named AN-51 (20). Binding studies of AN-51 to SSR-expressing tissues (for example, rat brain cortex, Dunning rat tumor R3327H, and MiaPaCa-2 pancreatic cancer cells) showed that AN-51 has only slight reduced SSR-binding affinity (2- to 10-fold) compared with its parental molecule RC-121 (20). In addition, a 27-d treatment of rats bearing MiaPaCa-2 pancreatic cancer xenografts with RC-121 (20 μg/d) and AN-51 (25 μg/d) resulted in a comparable suppression of serum growth hormone (GH) levels, suggesting that both SSR binding and biological activities are retained in the hybrid AN-51 molecule. More recently, Nagy et al. (21) developed a much more potent cytotoxic somatostatin analog, consisting of RC-121 coupled to 2-pyrrolino-doxorubicin (AN-201), which is a doxorubicin derivative that is 500–1,000 times more potent than doxorubicin. This novel molecule has been code-named AN-238 (Fig. 2). Compared with RC-121, the hybrid molecule AN-238 had a slightly reduced SSR binding affinity. Binding affinity, defined as 50% inhibitory concentration (IC50) value, the concentration of analog required to inhibit 50% of the specific binding of 125I-RC-160 somatostatin analog to SSR-expressing rat pituitary membranes, amounted to 0.31 nmol/L for RC-121 and 23.8 nmol/L for AN-238. This binding affinity of AN-238 was comparable to the binding affinities of several DTPA- and 1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid (DOTA)-coupled somatostatin analogs to human SST2 receptors (22). In superfused rat anterior pituitary cell cultures, it was shown that AN-238 fully preserved the GH-release inhibitory potency of RC-121 (21). Moreover, in vitro growth of gastric, breast, prostate, pancreatic, and SCLC cell lines was inhibited by AN-238 in a dose-dependent fashion with IC50-values ranging between 3.2 and 40 nmol/L, which were comparable to values of inhibition of cell growth by the 2-pyrrolino-doxorubicin alone (between 1.8 and 24 nmol/L). These data suggest that, like AN-51, AN-238 has retained the binding and functional properties of its parental molecules RC-121 and 2-pyrrolino-doxorubicin (21).

Structure of 3 different cytotoxic somatostatin analogs. (A) AN-238. Somatostatin analog RC-121 coupled to 2-pyrrolino-doxorubicin. (B) PNA-SST. Somatostatin analog coupled to antisense peptide nucleic acid (PNA). (C) Taxol-octreotide. Somatostatin analog octreotide conjugated to paclitaxel (taxol). Reprinted with permission from (21) and adapted from (25,26).
In addition to cytotoxic somatostatin analogs containing methotrexate, doxorubicin, or 2-pyrrolino-doxorubicin, other cytotoxic agents have been coupled to somatostatin analogs. Fuselier et al. (23) synthesized a series of somatostatin analogs coupled to the topoisomerase inhibitors camptothecin and combretastatin. Carbamates were used as linkers, because they are known to be relatively resistant in plasma, compared, for example, with ester-type linkages (24). One of these compounds, compound 2, had full retention of biologic activity compared with native somatostatin 14 (23). Compound 2 inhibited GH releasing hormone-stimulated GH release by cultured rat pituitary cells with an IC50 value in the low nanomolar range (0.27 nmol/L). In vitro cytotoxicity of this compound, as measured by a cell viability tetrazolium salt 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay was also in the nanomolar range (54.2 nmol/L) and comparable with inhibitory values described for AN-238. Huang et al. (25), showed some degree of efficacy of octreotide conjugated to taxol in MCF-7 cells. Octreotide-paclitaxel (Fig. 2) inhibited SSR-positive MCF-7 cell viability with an IC50 value of 100 nmol/L, whereas SSR-negative CHO cells showed a much lower sensitivity. Finally, a somatostatin analog conjugated to antisense peptide nucleic acids (PNA) targeting the n-myc oncogene showed retained functional activity in terms of GH suppression in vitro, as well as enhanced cytotoxicity to human SSR-expressing human IMR32 neuroblastoma cells, though with a relatively low efficacy (in the micromolar range) (26). It should be mentioned that stability of these compounds in vitro may be very important. If the compounds are degraded while measuring its cytotoxic effect, it may be possible that in cytotoxic assays also the effects of the cytotoxic compounds alone is involved. Thus the outcome of the in vitro studies does not necessarily reflect the potential of the compounds to selectively target tumor cells in vivo.
IN VIVO ANTITUMOR ACTIVITY OF CYTOTOXIC SOMATOSTATIN ANALOGS
Preclinical studies demonstrating that cytotoxic somatostatin analogs inhibit tumor growth in experimental tumor models are discussed in this section. In nude mice transplanted with the human MIA PaCa-2 pancreatic tumor, AN-51 significantly inhibited tumor growth, whereas methotrexate alone or RC-121 alone had no significant inhibitory effects (20), with methotrexate alone displaying a much higher toxicity compared with AN-51. No other preclinical studies with AN-51 have been identified in the literature. This is probably a result of the development of novel cytotoxic somatostatin analogs with enhanced cytotoxic activity. As indicated previously, AN-238 has a very high cytotoxicity in SSR-positive cells in vitro (21). In vivo, a large number of studies, mainly by Schally et al. (27–34) demonstrated that AN-238 has potent antitumor activity in proven SSR-positive experimental tumor models. Although these studies differ with respect to dose and number of administrations of AN-238, all studies clearly show that, compared with the cytotoxic parental molecule AN-201 (2-pyrrolino-doxorubicin), AN-238 displays a much higher antitumor activity and significantly lower toxicity. Using AN-238, effective tumor growth inhibition has been demonstrated in experimental rat and mouse models of human breast cancer, androgen-independent prostate cancer, ovarian cancer, SCLC and non–small cell lung cancer, pancreatic cancer, renal cell cancer, glioblastoma, colon cancer, and gastric carcinomas (27–40). In most studies, a much higher toxicity and lower or absent effectiveness on tumor growth was observed in animals treated with the cytotoxic radical AN-201 alone.
Because early degradation of cytotoxic somatostatin analogs in the circulation or in tissues may release the cytotoxic compound before it is able to reach its target (the SSR-positive tumor cell), it is important to demonstrate specificity of the effects of targeted cytotoxic somatostatin analogs. Several studies have addressed this issue and provide evidence for specificity of the effects of AN-238. Compared with the effect of AN-238, no or much lower antitumor activity is shown when the unlabeled somatostatin analog RC-121 and the cytotoxic compound are administered simultaneously (27,29,32,34,36). In some studies, no inhibitory effect of AN-238 could be demonstrated in SSR-negative tumors, whereas in the same studies SSR-positive tumor models showed an antitumor action of AN-238 (35,36,39). Pretreatment of animals with the unconjugated somatostatin analogs RC-160 or RC-121 could partially or completely prevent antitumor action by AN-238, suggesting that interaction with a specific SSR is required for AN-238 to exerts its action (36,38).
The mechanism through which AN-238 exerts its antitumor activity may include the induction of apoptosis and/or necrosis. An increase in the number of apoptotic cells, alone or in combination with an increase in necrotic areas in AN-238 treated tumors, has been shown in experimental human pancreatic tumors, gastric carcinomas, colon cancer, androgen-independent prostate cancer, SCLC, and non–small cell lung cancer (31,33,34,36,39,40).
SSR-targeted chemotherapy is effective in preclinical tumor models and may be a promising approach to treat SSR-positive tumors. Using SSR-targeted chemotherapy, lower dosages of the chemotherapeutic compound may be used, resulting in lower toxicity. Until now, however, no clinical trials have reported using cytotoxic LHRH, bombesin, or somatostatin analogs. In addition, evidence will have to be provided that cytotoxic somatostatin analogs can be internalized by SSR-positive tumor cells. As for the concept of SSR-targeted radiotherapy, the efficacy of SSR-targeted chemotherapy will be determined by the SSR expression level on tumor cells, the capability of the cytotoxic somatostatin analog to selectively bind with high affinity to these SSRs, the stability of the compound in circulation, and the amount of internalization of the cytotoxic radicals by tumor cells. Moreover, in contrast to the use of radiolabeled somatostatin analogs coupled with therapeutic radionuclides emitting β-particles with long particle range (such as 90Y and 177Lu, where radiation emitted from receptor-positive tumor cells may kill receptor-negative cells in tumors with receptor heterogeneity), it is expected that targeted cytotoxic somatostatin analogs require a more homogenous SSR expression by tumors to exert their action (41). However, it may be hypothesized that after the induction of necrosis a local release of the cytotoxic radical may induce a “bystander effect” on adjacent SSR-negative cells (Fig. 1). In this respect it is important to note that some human cancers, such as breast cancer, may have a nonhomogenous intratumor SSR expression and that several experimental human tumor models express SSR, whereas studies in primary human tumors, such as non–small cell lung cancer, colorectal cancer, and exocrine pancreatic cancer, have shown a lack of SSR at the protein level (3).
SIDE EFFECTS OF CYTOTOXIC SOMATOSTATIN ANALOGS
It is well known that SSRs are widely expressed in various tissues, including rapidly proliferating cells in the gastrointestinal tract. Therefore, some side effects may occur with the use of cytotoxic somatostatin analogs. At present, the toxicity of cytoxic somatostatin analogs, in particular AN-238, has been evaluated only in rats and mice. These studies do not show the specific toxicity of AN-238 in the gastrointestinal tract, pituitary, or kidneys. No significant changes in pituitary function, as measured by GH and luteinizing hormone levels, have been observed in nude mice bearing human breast cancer xenografts and treated with a single intravenous injection of 250 nmol/kg AN-238 and followed for 60 d (27). In the majority of in vivo studies in experimental tumor models a certain degree of toxicity of AN-238 treatment as measured by animal deaths, loss of body weight, and leukopenia has been reported. All studies clearly showed that toxicity of AN-201 in this respect was much greater than that of AN-238 treatment (1,2). An important factor determining the toxicity of cytotoxic somatostatin analogs is their stability. The intact molecules should be able to reach their target tumor cells, bind to SSRs, internalize, and exert their cytotoxic action locally. AN-201 is linked to the RC-121 somatostatin analog by an ester bond. Therefore, deconjugation by nonspecific esterases may occur. Differences between mice and rats with respect to toxicity of AN-201 and AN-238 have been reported and could be the result of different esterase activities between the species (21). The half-life of the ester bond is about 20 min in nude mice, 1 h in rats, and about 2 h in human serum (33,42). In addition, mice with suppressed esterase activity tolerated up to 3 times higher doses of AN-238 than AN-201 (1). In this respect, the development of novel cytotoxic somatostatin analogs using carbamates as potential linkers, which are known to be relatively resistant in plasma, is of specific interest (23). One of these compounds, the camptothecin conjugate 2, has a half-life of 18 h in rat serum.
RGD-SOMATOSTATIN ANALOGS
RGD
Integrins are a large family of heterodimeric glycoprotein cell surface receptors that regulate cell–cell and cell–matrix interactions. They are composed of 2 transmembrane glycoproteins, α- and β-subunits, and each αβ-combination has its own binding specificity. These integrin receptors mediate a variety of cell adhesion events and signal transduction processes and are involved in an array of pathologic events, such as tumor metastasis, angiogenesis, thrombosis, and osteoporosis. One of these integrins, the αvβ3 receptor, is able to bind several extracellular matrix proteins via the sequence of 3 amino acids: arginine (R), glycine (G), and aspartate (D), commonly called RGD, after the single-letter codes assigned to these amino acids (43). The αvβ3 receptor is expressed on various malignant human tumors and is highly expressed in neovascular endothelial cells, which are actively involved in angiogenesis (44). Based on the RGD sequence, several small, highly active peptides have been designed to antagonize the αvβ3 receptor. These are synthetic RGD molecules that bind with high affinity to this receptor and have been shown to be inhibitors of cell adhesion, migration, growth, and differentiation (45–48). These agents also inhibit the growth of new blood vessels and induce tumor regression in animal models, presumably by restriction of the blood supply to the tumor (49–52).
In addition to the RGD binding sites found on integrins, it has been shown that several members of the procaspase family of apoptosis genes contain such binding motifs as well. Procaspase-1 and -3 contain potential RGD-binding motifs near the site necessary for activation of the procaspases (53). Caspases are a small group of specific cysteine proteases that are key factors in programmed cell death (54). These intracellular factors exist as latent zymogens, which, when activated, induce orderly cell death. Buckley et al. (53) first demonstrated that RGD peptides are able to directly activate caspase-3 and induce apoptosis in cells by a mechanism that is completely independent of their association with integrin receptors. Additional work by others has shown that molecules specific for glycoprotein IIb/IIIa integrins can also stimulate caspase-3 activity (55). Because caspase-3 is one of the key executioner proteases in the apoptosis pathway, it seems likely that this enzyme will be an important site of action for targeted therapeutics designed to selectively induce cell death (54). In a recent study the effects of an RGD-containing peptide (Arg-Gly-Asp-Ser [RGDS]) was investigated in human umbilical vein endothelial cells (56). It was shown that after 4 h of incubation both caspase-8 and caspase-9 were activated, and caspase-3 was activated after 24-h incubation with RGDS and, thereby, induced apoptosis. These findings all support the hypothesis that RGD peptides may directly trigger the caspase cascade at an early level. Therefore, direct RGD interaction with caspases and their activation at different levels may represent a novel mechanism of apoptosis induction, in addition to the known death receptor- and mitochondria-mediated pathways.
In addition to cytotoxic RGD analogs, radionuclide-coupled RGD have been designed for imaging and radiotreatment (57,58). These analogs have a high affinity to neovascular endothelium and tumors. Specific uptake and retention of radioactivity in a transplanted tumor has been described in an animal model (58). These analogs are promising in the targeted radiotreatment of tumor vascularization. A new and interesting approach is the use of chimeric peptides. It has been shown that RGD-containing peptides can be used for homing to tumors via the αvβ3 receptors, which are expressed on tumor vasculature (59,60). These peptides have the potential to allow targeting of tumor vasculature with toxins as proapoptotic peptides (52). A study with a fusion protein containing the RGD sequence and the Fc fragment of mouse IgG (RGD/mFc) was designed to target the Fc portion of IgG to the tumor vasculature to elicit an antiangiogenesis immune response (61). The Fc fragment can trigger phagocytosis by neutrophils or macrophages. The Fc fragment is also able to activate natural killer cells to lyse the cells bearing the Fc fragment. The researchers used the Fc fragment of IgG that triggers the cascade of the complement system. It was shown that tumor angiogenesis and tumor growth could be inhibited after treatment with RGD/mFc.
RGD Somatostatin
Another fascinating approach is the use of RGD peptides as an apoptosis-inducing agent. Using a Pioneer (Applied Biosystems) synthesizer and an Fmoc strategy, we synthesized via solid-phase peptide synthesis a new peptide, RGD-DTPA-octreotate (c[Arg-Gly-Asp-d-Tyr-Asp]-Lys[DTPA]-Phe-c[Cys-Tyr-d-Trp-Lys-Thr-Cys]-Thr). The action of this novel compound is based on the overexpression of SST2 on tumor cells. RGD-DTPA-octreotate is composed of the SSR-targeting peptide Tyr3-octreotate, the chelator DTPA to enable radiolabeling, and the apoptosis-inducing RGD sequence. We choose to create a cyclic RGD derivative, because it was found that cyclic derivatives of RGD peptides were more than 100-fold better when compared with linear variants (46).
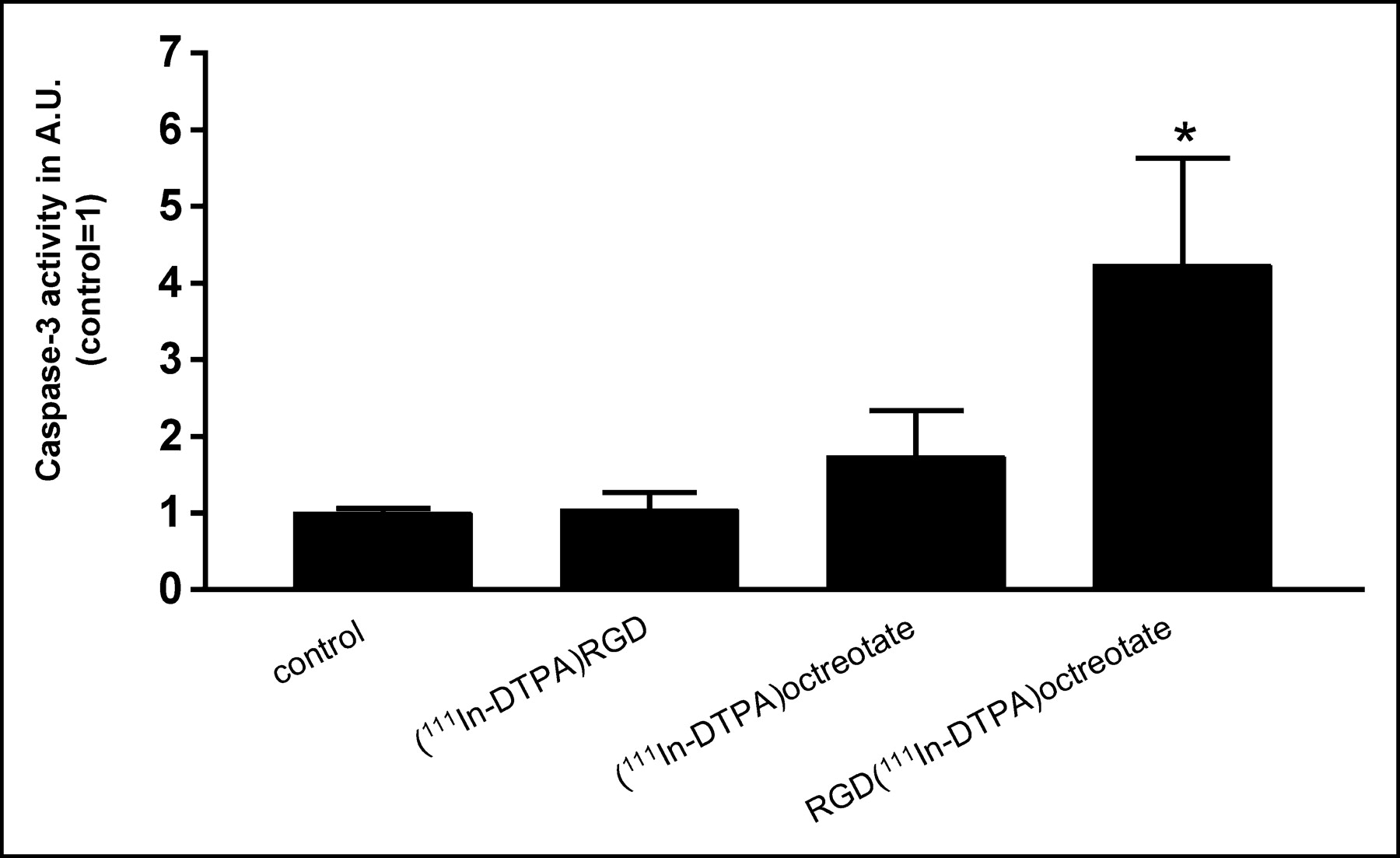
This hybrid peptide RGD-DTPA-octreotate has a high affinity for SST2 and internalizes in vivo and in vitro into tumor cells, mostly via SST2, most probably because of the higher affinity of octreotate for SST2 than that of RGD for the αvβ3-receptor (62). Preliminary results showed that the 111In-labeled compound had a pronounced tumoricidal effect in a clonogenic assay in comparison with the 2 monopeptides RGD and Tyr3-octreotate. Furthermore, 111In-labeled RGD-DTPA-octreotate showed a clear increase in caspase-3 levels, with the highest levels found after 24-h incubation (63) (Fig. 3). From these results it is clear that the 111In-labeled peptide RGD-DTPA-octreotate is more powerful in promoting apoptosis than the 2 monopeptides RGD and octreotate. The radiolabeled hybrid peptide, therefore, can significantly enhance the therapeutic efficacy of somatostatin-based agents. To investigate the contribution of the radionuclide, unlabeled RGD-DTPA-octreotate was tested as well. It was shown that the unlabeled compound RGD-DTPA-octreotate also gives an increase in caspase-3 levels after 24-h incubation. This makes it interesting to consider the use of the unlabeled hybrid peptide RGD-octreotate as a novel treatment as well.

Caspase-3 activity after incubation with 111In-labeled RGD [c(Arg-Gly-Asp-d-Tyr-Asp)]-octreotate, or RGD-octreotate [c(Arg-Gly-Asp-d-Tyr-Asp)-Lys(DTPA)-Phe-c(Cys-Tyr-d-Trp-Lys-Thr-Cys)-Thr] in the CA20948 cell line. Caspase-3 activity was measured after 24 h, expressed in arbitrary units (AU), control set on 1. Experiments were performed in triplicate, and each sample was measured in triplicate. Bars represent mean ± SEM; *P < 0.05 versus control.
The mechanism of action of this hybrid peptide RGD-DTPA-octreotate could be explained with the hypothesis that RGD peptides may directly trigger the caspase cascade at an early level. Because a clear increase in caspase-3 levels is found after 24 h, it is possible that other caspases, such as caspase-8 and -9, could be activated first, as reported previously (56). We cannot rule out the contribution of the chelator DTPA in the active mechanism of this hybrid peptide. It could play a role, for example, in resistance to degradation or as a scavenger inside the cell. In addition to the hypothesis that the RGD sequence activated the caspase cascade, RGD-DTPA-octreotate could activate the known apoptotic pathway, the mitochondrial-mediated pathway, which also leads to an increase in caspase-3 levels (Fig. 4). Clearly more research must be performed to understand the precise action of RGD-DTPA-octreotate. The development of hybrid molecules that combine targeting and an effector function, such as apoptosis, can be a new approach in the treatment of cancer and inflammatory diseases.
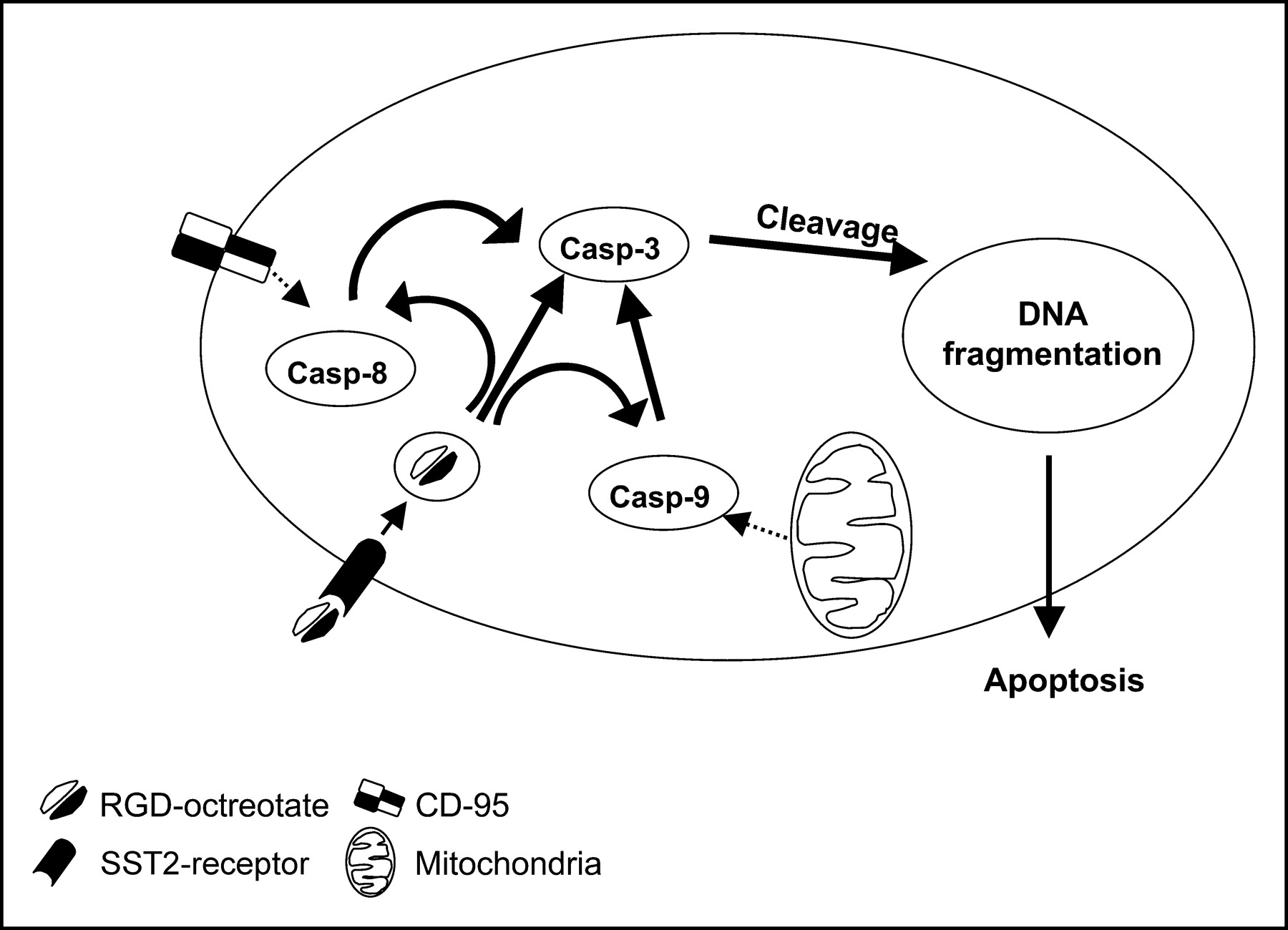
Possible mechanism(s) of action of hybrid peptide RGD-octreotate.
CONCLUSIONS
Several cytotoxic somatostatin analogs have been developed in the past decade, including analogs coupled to chemotherapeutic agents, taxol, antisense peptide nucleic acids, and RGD. Effectiveness as antitumor agents has been demonstrated in in vitro studies, as well as in in vivo experimental tumor models. The capability of such hybrid molecules to target SSR-positive tumor cells in vivo will very much depend on their stability in the circulation, their SSR (subtype) binding affinity, as well as the capability of SSR-positive tumor cells to internalize such hybrid molecules. Nevertheless, current preclinical data clearly provide evidence for successful targeting of SSR-positive tumors in vivo using somatostatin analogs coupled to chemotherapeutic agents, which have increased efficacy and considerable lower toxicity compared with unconjugated cytotoxic radicals, as well as for in vitro tumor cell targeting using taxol-, antisense oligo-, and radiolabeled RGD-coupled somatostatin analogs.
Footnotes
Received July 5, 2004; revision accepted Oct. 21, 2004.
For correspondence or reprints contact: Leo J. Hofland, PhD, Department of Endocrinology, Room Ee585c, Erasmus MC, Dr. Molewaterplein 50, 015 GE Rotterdam, The Netherlands.
E-mail: l.hofland{at}erasmusmc.nl
REFERENCES
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- SOMATOSTATIN RECEPTOR SUBTYPE EXPRESSION IN TUMOR TISSUES
- INTERNALIZATION OF SOMATOSTATIN RECEPTOR SUBTYPES
- DEVELOPMENT AND IN VITRO CHARACTERIZATION OF CYTOTOXIC SOMATOSTATIN ANALOGS
- IN VIVO ANTITUMOR ACTIVITY OF CYTOTOXIC SOMATOSTATIN ANALOGS
- SIDE EFFECTS OF CYTOTOXIC SOMATOSTATIN ANALOGS
- RGD-SOMATOSTATIN ANALOGS
- CONCLUSIONS
- Footnotes
- REFERENCES
- Figures & Data
- Info & Metrics
Related Articles
Cited By...
- No citing articles found.