


Abstract
Auger electron–emitting radionuclides have potential for the therapy of small-size cancers because of their high level of cytotoxicity, low-energy, high linear energy transfer, and short-range biologic effectiveness. Biologic effects are critically dependent on the subcellular (and even subnuclear) localization of these radionuclides. Our goals were the design, synthesis, and in vitro preclinical assessment of new trifunctional conjugates of somatostatin that should aim at the nucleus and, therefore, ensure a longer retention time in the cell, a close approximation to the DNA, and the success of Auger electron emitters in targeted radionuclide therapy as well as also improve other targeted therapy strategies. Methods: Three trifunctional derivatives of [(1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid)0,Tyr3]octreotide (DOTA-TOC) bearing the nuclear localization signal (NLS) (of simian virus 40 large-T antigen) PKKKRKV in 3 different positions relative to the somatostatin analog sequence were synthesized using solid and solution phase peptide synthesis. These compounds together with DOTA-TOC and DOTA-NLS derivatives were labeled with 111In and tested for binding affinity, internalization, externalization, and nuclei localization on AR4-2J cells and on human embryonic cells stably transfected with sst2A. Results: The two N-terminal derivatives preserved the sstr2A binding affinity. Their rate of internalization in all tested sstr-expressing cell lines was always superior for the trifunctional derivatives in comparison with the parent compound. A 6-fold increase in cellular retention from the total internalized activity and a 45-fold higher accumulation in the cell nuclei were found for one of the N-terminally modified compounds compared with [111In]-DOTA-TOC. The C-terminal conjugate was inferior in all tests compared with the parent compound. Conclusion: These encouraging results support our hypothesis that an additional NLS sequence to the DOTA-TOC could not only provide a better carrier for Auger electron–emitting radionuclides but also ensure a longer radioactivity retention time in the tumor cell.
Given the overexpression of somatostatin receptors in a variety of neuroendocrine tumors and their metastases (1), peptide receptor–mediated radiotargeted therapy (PRRT) using radiolabeled somatostatin derivatives is the method of choice for their treatment. The gold standard in our clinic is represented by DOTA-TOC (DOTA = 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid; TOC = Tyr3-octreotide), a radiopeptide that, labeled with 90Y (2), has now almost a decade of clinical experience in therapy (3). 90Y is a β-particle emitter with a maximum electron energy of 2.3 MeV and an optimal range in tissue of up to 42 mm, its cytotoxicity being primarily due to the crossfire effect (4). However, 90Y seems less suitable for the therapy of small metastases or disseminated single tumor cells, because very small tumors will not be able to absorb all electron energy emitted by 90Y in the tumor cells (5). In very small lesions, PRRT with Auger electron–emitting radiopharmaceuticals may be a better choice. Auger electron emitters decaying in the neighborhood of the DNA produce similar amounts of reactive chemical radical species as do α-emitters, which are regarded as the classical form of high linear energy transfer (LET) radiation (6).
Several encouraging accounts on radiotherapy using internalizing antibodies conjugated to radionuclides emitting low-energy electrons have been recently published (7–9). Accompanied by studies on comparison of the usefulness of Auger electron emitters versus β-particle emitters in radioimmunotherapy (6,10,11), these reports demonstrate the superior cell killing efficacy of Auger electron emitters in small tumors. Also, [111In-DTPA]octreotide (DTPA = diethylenetriaminepentaacetic acid) has already been used for radionuclide therapy in patients with somatostatin receptor–positive tumors (12). An additional advantage of using low-energy emitters such as 111In is the lack of renal toxicity. It was shown that the kidney is the major critical organ for PRRT with 90Y-DOTA-TOC (13), whereas the use of [111In-DTPA]octreotide did not show any renal toxicity even though calculated kidney doses were higher than 40 Gy (12). Along with the in vitro therapy investigations performed by Capello et al. (14), these studies show that the usefulness of this type of PRRT using low-energy emitters is clearly dependent on the high accumulation of the radioligand in the tumor cell. Moreover, Tiensuu Janson et al. (15) have shown that the clinical effectiveness of [111In-DTPA]octreotide could be explained by the translocation of 111In to the perinuclear and nuclear area of the cell. Consequently, enhancing the tumor uptake and retention of radiolabeled somatostatin analogs could provide more effective in situ radiotherapy, optimizing the energy transfer from Auger electron emitters to tumor DNA (16).
In this respect we reasoned that a new functional unit for nucleus targeting and prolonged cell retention could be added to the DOTA-TOC conjugate. For this we have chosen the classical nuclear localization signal (NLS) of the simian virus 40 large-T antigen, the heptapeptide H-Pro-Lys-Lys-Lys-Arg-Lys-Val-OH (17). This sequence serves as a tag to proteins, indicating their destination to the cell nucleus and assisting in the transport through the nuclear membrane. To function properly, the NLS conjugates must be located in the cytoplasm, but they are not readily incorporated into cells (18). We synthesized previously 3 such derivatives bearing the NLS unit in 3 different positions relative to the somatostatin analog sequence (19). In this study we investigated the biologic in vitro outcome of these NLS-DOTA-TOC derivatives (1), (2), and (3) (Fig. 1) labeled with 111In, by means of receptor-binding affinity, rate of internalization, cellular retention, and cellular nuclear uptake. This evaluation was done in comparison with the parent compound, 111In-DOTA-TOC, but also with the 111In-DOTA-Ahx-Pro-Lys-Lys-Lys-Arg-Lys-Val-OH (4) derivative, which served as a control (Ahx = aminohexanoic acid).

Structures of DOTA-TOC and of NLS-conjugates (1), (2), (3), and (4).
MATERIALS AND METHODS
Radiolabeled Peptide Derivatives
DOTA-TOC and the conjugates (1), (2), and (3) were synthesized as previously described (19). Also, DOTA-Ahx-Pro-Lys-Lys-Lys-Arg-Lys-Val-OH (4) was synthesized as recently reported (19). DOTA was coupled as a tris(t-butyl ester) using the monoamide approach. 111InCl3 was obtained from Mallinckrodt Medical. All of the conjugates were labeled with 111InCl3 as described (20).
Cell Culture
The AR4-2J cell line was maintained by serial passage in mono-layers in Dulbecco’s modified Eagle medium (DMEM; Cambrex Bio Science), supplemented with 10% fetal bovine serum, amino acids, vitamins, and penicillin–streptomycin, in a humidified 5% CO2 atmosphere at 37°C. Human embryonic kidney (HEK) 293 cells stably expressing sstr2A receptors were transfected as described (21) and were grown in DMEM supplemented with 10% fetal bovine serum, penicillin–streptomycin, and G418 (500 μg/mL) in a humidified 5% CO2 atmosphere at 37°C.
Saturation Binding Assays
HEK cells stably expressing sstr2A were grown as described previously (21). All culture reagents were supplied by BioConcept. For each of the tested compounds, saturation binding experiments on intact cells were performed, using increasing concentrations of the 111/natIn-DOTA-peptide ranging from 0.1 to 1,000 nmol/L. 111/natIn-DOTA-TOC was run in parallel as a control using the same increasing concentrations. natIn-DOTA-TOC at 1 μmol/L was used to quantify the nonspecific binding. Cells were seeded near confluence into 6-well plates using the growth medium and incubated overnight. The medium was removed on the next day, binding buffer (DMEM with 1% fetal bovine serum, pH 7.4) was added to the wells, and the cells were incubated for 1 h at 37°C. For each radioligand, triplicates were prepared for every concentration, for both total binding and nonspecific binding. Before adding the radioligands to the wells, the plates were placed on ice for 30 min. After adding the radioligands and natIn-DOTA-TOC for nonspecific binding, the plates were incubated for 2 h at 4°C. After this interval, the binding buffer was aspirated and the cells were washed twice with ice-cold phosphate-buffered saline (PBS, pH 7.4); this represented the free fraction. Finally, the cells were collected with 1N NaOH; this corresponded to the bound fraction. A control for each radioligand was prepared to check the amount of conjugate that internalizes at 4°C. The radioactivity in the free and bound fractions was measured using a γ-counter (Cobra II; Packard Instrument Co.). Specific binding was calculated as the difference between the radioactive levels without versus with 1 μmol/L natIn-DOTA-TOC. Dissociation constant (Kd) values were calculated from Scatchard plots of the obtained data using Origin 5.0. software (Microcal Software, Inc.).
Radioligand Internalization Studies
The apparatus and procedures for the cell internalization experiments are based on previously described methods (20). Briefly, for all cell experiments, the cells were seeded at a density of 0.8–1.1 million cells per well in 6-well plates and incubated overnight with internalization buffer to obtain good cell adherence. When different radiolabeled peptides were compared in cell experiments, the same cell suspension–containing plates were used. Furthermore, the internalization rate was linearly corrected to 1 million cells per well in all cell experiments. Medium was removed from the 6-well plates and cells were washed once with 2 mL of internalization buffer (DMEM, 1% fetal bovine serum, amino acids, and vitamins, pH 7.4). Next, 1.5 mL of internalization buffer were added to each well and incubated at 37°C for about 1 h. Thereafter, approximately 500,000 cpm or 0.02 MBq of 111/natIn-labeled peptides per well (2.5 pmol/well) to a final concentration of 1.67 nmol/L were added to the medium and the cells were incubated at 37°C for the indicated time periods in triplicates. To determine nonspecific membrane binding and internalization, cells were incubated with radioligand in the presence of 1 μmol/L natIn-DOTA-TOC. Cellular uptake was stopped by removing medium from the cells and by washing twice with 1 mL of ice-cold PBS. Acid wash for 10 min with a pH 2.8 glycine buffer on ice was also performed twice. This procedure was performed to distinguish between membrane-bound (acid releasable) and internalized (acid resistant) radioligand. Finally, the cells were treated with 1N NaOH. The culture medium, the receptor-bound fraction, and the internalized fraction were measured radiometrically in a γ-counter (Cobra II).
Cellular Retention Studies
For cellular retention studies, HEK cells sstr2 stably transfected (1 million) were incubated with 2.5 pmol per well (1.67 nmol/L) of 111/natIn-labeled DOTA-peptide for 120 min; then the medium was removed and the wells were washed twice with 1 mL of ice-cold PBS. In each experiment, an acid wash for 5 min on ice with a pH 2.8 glycine buffer was performed twice to remove the receptor-bound ligand and a PBS wash was performed quickly afterward to restore the physiologic pH. Cells were then incubated again at 37°C with fresh internalization buffer (DMEM containing 1% fetal bovine serum, pH 7.4). After different time points, the external medium was removed for quantification of radioactivity in a γ-counter and replaced with fresh 37°C medium. The cells were solubilized in 1N NaOH and removed, and the internalized radioactivity was quantified in a γ-counter. The recycled fraction was expressed as the percentage of the total internalized amount per 1 million cells.
Nuclei Isolation Assay
A nuclei isolation kit (Nuclei EZ Prep Kit; Sigma-Aldrich Chemie Gmbh) was used to separate and quantify the amount of radioactivity stored in the nuclei. The preparation was done according to the manufacturer’s instructions. HEK cells stably expressing sstr2 were seeded in Petri dishes at a density of 18–20 million per dish with growth medium and incubated overnight at 37°C. On the next day the medium was removed, the cells were washed twice with PBS, and internalization medium was added to the dishes (DMEM, 1% fetal bovine serum, amino acids, and vitamins, pH 7.4). The cells were incubated at 37°C for 1 h. The conjugates (3) and DOTA-TOC were radiolabeled with 111In at a specific activity of 37 GBq/μmol. Fifty picomoles from each radioconjugate were added to the cells, followed by incubation at 37°C for 4 h (triplicates were used for each compound). After this interval, the dishes were placed on ice, the media were aspirated, and the plates were washed 4 times with ice-cold PBS. Subsequently, the cells were harvested and lysed according to the Nuclei EZ Prep Kit manufacturer’s instructions. The purity of the final nuclei was determined by careful visual microscopic inspection of the nuclei diluted in trypan blue counting solution (Fluka Chemie Gmbh), using a hemacytometer. The radioactivity in the nuclei and the cytoplasmatic fractions collected for each of the 2 compounds was quantified in the γ-counter.
Statistical Methods
The Student t test was used to determine statistical significance. Differences at the 95% confidence level (P < 0.05) were considered significant.
RESULTS
Radiolabeled Peptide Derivatives
The structures of the investigated derivatives are shown in Figure 1. Following the same procedure as for (1), (2), (3), and DOTA-TOC (19), compound (4) was synthesized to be used as a negative control for the in vitro studies described herein. The efficient radiolabeling of these derivatives with 111In was confirmed by high-performance liquid chromatography equipped with a γ-detector (data not shown).
Binding Affinity Profiles
Table 1 shows the Kd values of the radiopeptides studied in this work as their 111/natIn-complexed versions in comparison with 111/natIn-DOTA-TOC for the somatostatin receptor subtype 2. The values were obtained by performing saturation binding experiments on intact cells expressing sstr2A. Except for 111/natIn-(4), all synthesized derivatives show specific binding affinity to this receptor. Whereas the two N-terminally modified DOTA-TOC derivatives, 111/natIn-(2) and 111/natIn-(3), maintain a good affinity to this receptor subtype with values comparable to those of 111/natIn-DOTA-TOC, the C-terminally modified DOTA-TOC derivative (1) labeled with 111/natIn displays a 100-fold drop in the binding affinity.
Binding Affinities (Kd ± SD, nmol/L; n = 3) of NLS-Derivatives and DOTA-TOC Labeled with 111In for sstr2A
In Vitro Internalization Studies
Figure 2 displays the rates of internalization of all 5 111In-labeled compounds in 2 cell lines: AR4-2J rat pancreatic tumor cell line (Fig. 2A) and HEK cells stably expressing sstr2A (Fig. 2B). In both cases, the highest rate of internalization at 4 h corresponds to 111In-(3) (22.2% in AR4-2J and 39.9% in HEK-sstr2). The control performed with 111In-(4) shows no uptake in any of the cell lines used. At 4 h, 111In-(2) and 111In-(3) have a 3-fold and a 3.6-fold, respectively, higher uptake in AR4-2J compared with 111In-DOTA-TOC. Correlating with the affinity profile, 111In-(1) reveals a significant decrease in the internalization rate in AR4-2J, compared with the parent compound (2.2% vs. 6.0%). The same order of uptake at 4 h is maintained in HEK cells expressing sstr2, but with higher absolute values for all of the somatostatin-based derivatives investigated. In this cell line, the 30-min and 1-h uptake of 111In-(3) is lower than that for 111In-(2), but after a 2-h incubation both compounds have similar cellular uptakes. At 4 h, 111In-(3) has a significantly higher accumulation than that for 111In-(2) (P < 0.001). Blocking studies were performed at all time points in both cell lines with 1 μmol/L natIn-DOTA-TOC, demonstrating that internalization was receptor mediated.
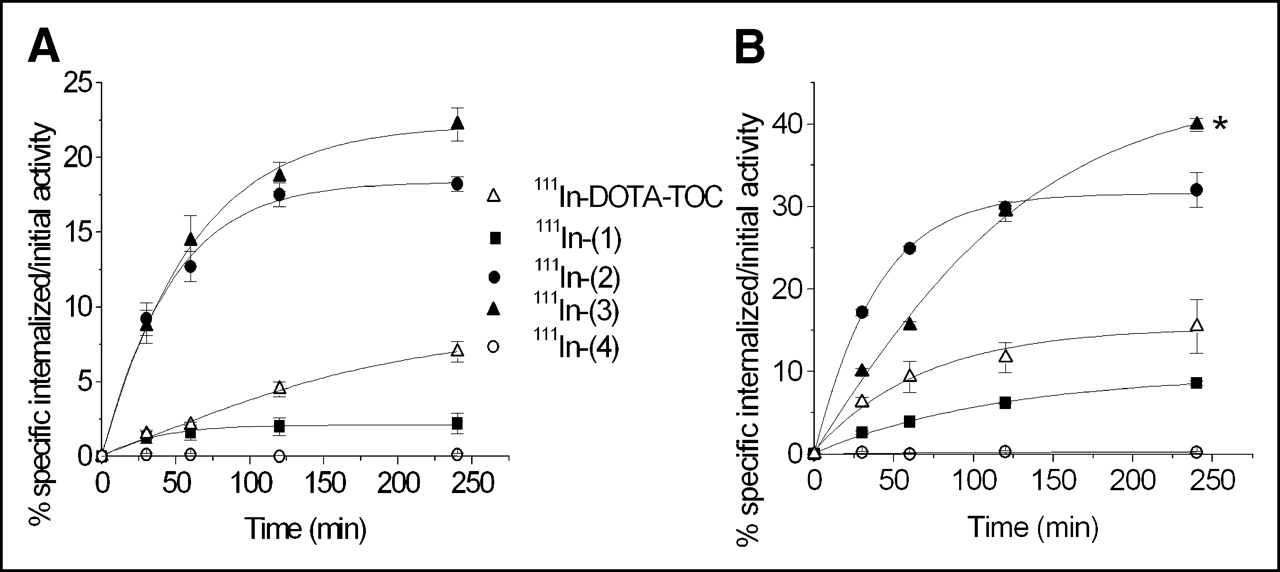
Comparison of internalization rates of 111In-labeled conjugates (1), (2), (3), and (4) and DOTA-TOC in AR4-2J cells (A) and in sstr2A-HEK cells (B). Values represent specific internalization (% initial activity to 1 million cells at 1.67 nmol/L concentration) and are results of 3 independent experiments with triplicates in each experiment. *P < 0.001.
In the same internalization experiment we collected the membrane surface-bound radioligand performing twice acid washes with pH 2.8 glycine buffer before the treatment with 1N NaOH for each compound. The results are shown in Figure 3. No surface-bound radioligand was found for 111In-(4) in both cell lines. Very low amounts of cell-surface–associated activity were also recovered for 111In-DOTA-TOC. In contrast, all 3 NLS-derivatives of DOTA-TOC exhibit relatively high amounts of surface-bound radioactivity in both cell lines, in the order 111In-(3) > 111In-(2) > 111In-(1). The pattern of the percentage of surface-bound radioligand of the total amount (2.5 pmol/well) is the same for all compounds, including 111In-DOTA-TOC: a rapid increase within 30 min and a descent from 1 to 4 h.
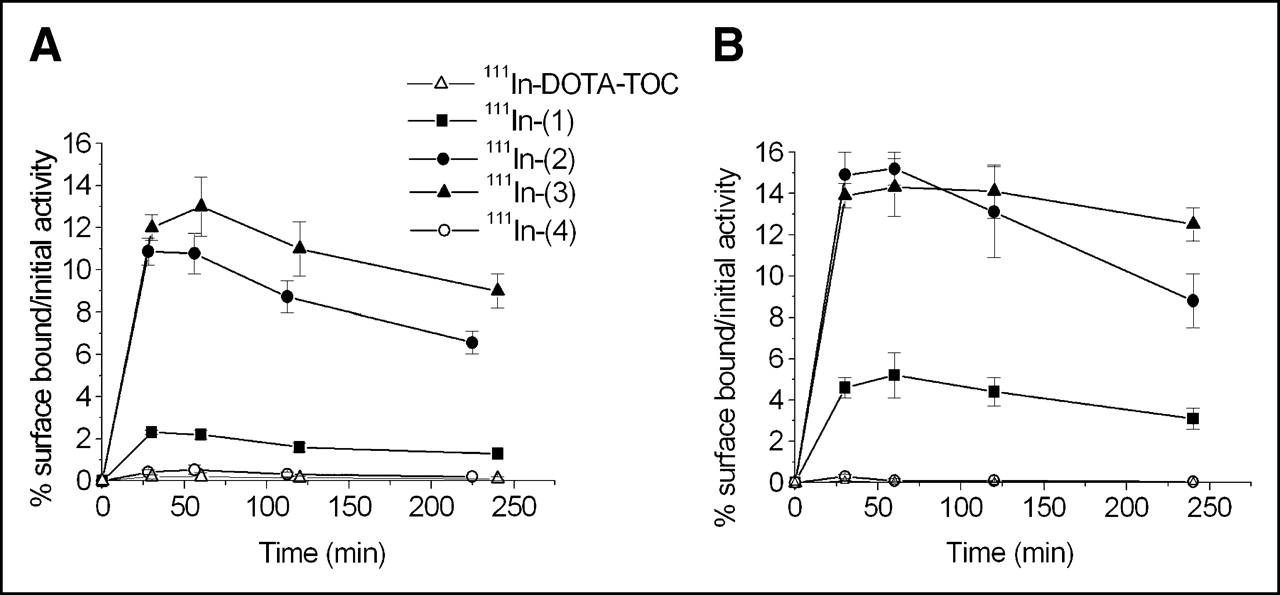
Comparison of surface-bound amounts of 111In-labeled conjugates (1), (2), (3), and (4) and DOTA-TOC in AR4-2J cells (A) and in sstr2A-HEK cells (B). Values represent specific binding (% initial activity to 1 million cells at 1.67 nmol/L concentration) and are results of 3 independent experiments with triplicates in each experiment.
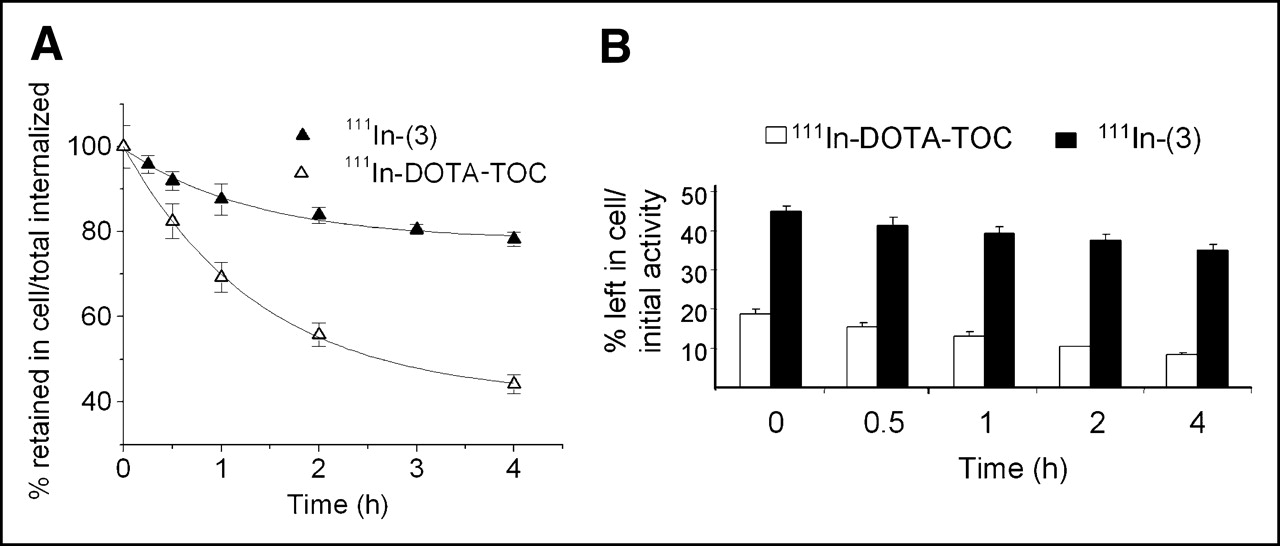
Cellular Retention of 111In-(3) and 111In-DOTA-TOC in HEK Cells Expressing sstr2A
Cellular retentions of 111In-(3) and 111In-DOTA-TOC were analyzed and compared in HEK cells expressing sstr2A. In these experiments, the radiopeptides were allowed to internalize for 120 min; cells were then washed twice with PBS before removing the receptor-bound ligand with glycine buffer, pH 2.8. Warm medium (37°C) was then added and removed after 15, 30, 60, 120, and 240 min and measured for radioactivity. Figure 4A illustrates the cellular radioactivity retention of these compounds over time, expressed as the percentage left in the cell from the total amount internalized. As 111In-(3) seems to reach a plateau after 4 h, 111In-DOTA-TOC continues to externalize. Almost 80% from the 2-h internalized 111In-(3) is still in the cells after 4 h, whereas only 50% of the 111In-DOTA-TOC 2-h uptake is still maintained at this interval. This difference becomes more prominent if the retention in the cell is expressed as the percentage from the total amount of radiopeptide added to the wells (2.5 pmol/well/million cells), as shown in Figure 4B. Thus, after 2 h of internalization, followed by 4 h of externalization, 35% of the NLS-derivative is still retained in the cells (equivalent with 0.88 pmol/well/million cells), whereas only 8.2% of the 111In-DOTA-TOC added remained intracellular (equivalent with 0.2 pmol/well/million cells).
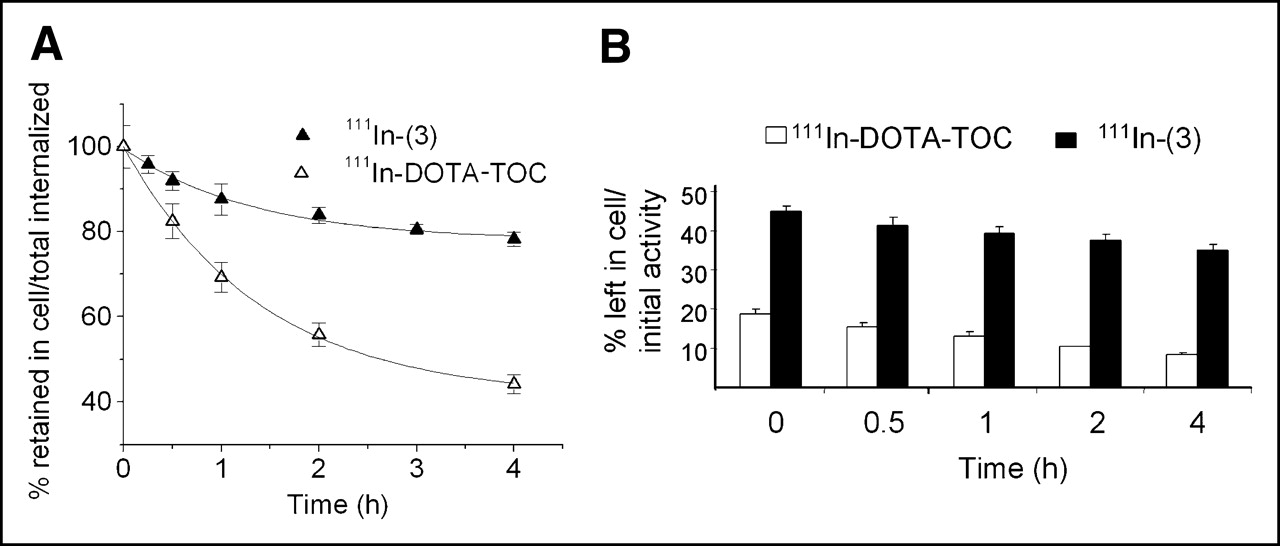
Comparison of cellular retention over time between 111In-(3) and 111In-DOTA-TOC in sstr2A-HEK cells at 37°C after 2 h of internalization. Values are expressed as percentage retained in cell from total amount internalized (A) and from total amount of added radioligand (B).
Uptake of 111In-(3) and 111In-DOTA-TOC in Nuclei of HEK Cells Expressing sstr2A
The isolation of HEK-sstr2 nuclei using the Nuclei EZ Prep Kit yielded high- purity nuclei, as revealed by the trypan blue staining test. This experiment was done to determine and compare the amount of radioactivity stored in nuclei after 1-, 4-, and 24-h continuous incubation of 111In-(3) and 111In-DOTA-TOC at 37°C with HEK cells expressing sstr2A. The results are shown in Figure 5 and Table 2. Thus, independently of the unit of measure used to express this outcome, there is a massive difference between the nuclear uptakes of the 2 radioligands especially at 1 and 4 h. If the total internalized radioactivity is taken as reference, then 111In-(3) has a 15-times higher nuclear accumulation at 1 h compared with that of the parent compound and the difference becomes even larger at 4 h with an 82-fold increase in the percentage in the nuclei in favor of 111In-(3) (Fig. 5). Reported as the percentage of the added radioligand or the amount of radiopeptide added (Table 2), the gap between the nuclear uptake of the 2 conjugates goes from 20-fold at 1 h to 45-fold at 4 h and a 16-fold difference at 24 h, always in favor of 111In-(3).

Percentage of nuclear uptake of radioactivity in sstr2A-HEK cells incubated with 111In-(3) and 111In-DOTA-TOC for 1, 4, and 24 h. Values represent percentage from total amount internalized.
Comparison of Cell Nuclear Uptake Between 111In-(3) and 111In-DOTA-TOC in HEK Cells Stably Expressing sstr2A
DISCUSSION
The design of effective Auger electron–or low-energy electron–emitting targeting agents for in vivo targeting and treatment of cancers becomes of increasing interest. Neglected initially for therapeutic purposes because of their low-energy and consequent short range, Auger electron cascades are now being seriously considered. The majority of low-energy Auger electrons emitted during radioactive decay deposit their energy over subcellular dimensions, producing highly localized energy density in the immediate vicinity of the decay site (22). In vivo and in vitro studies demonstrate that the toxicity of Auger electron emitters approximates that for low-LET radiation when the emitter is localized on the membrane or in the cytoplasm and that for high-LET α-particles when localized in the nucleus or in its proximity (23). Recently, the hypothesis that the radiotoxicity of Auger electrons is caused only by direct ionization of the DNA has been proven inaccurate. Apparently 90% of Auger electrons’ toxicity is due primarily to indirect mechanisms (24,25). Lacking the crossfire effect, for a long time it was assumed that the toxicity and therapeutic potential of low-energy electron emitters requires the radiotargeting of each and every tumor cell. This concept has been recently established as inexact, as the decay of such isotopes leads to a so-called “bystander effect,” proven by the in vivo use of 125I-deoxyuridine (26). This is translated into an in vivo inhibition or retardation of tumor growth in nonradiotargeted cells by signals produced in Auger electron–labeled cells. Nevertheless, a long enough time of retention in the targeted cells and intranuclear location of the radioligand would increase the potential success of this type of therapy (27).
On the basis of previous work reporting targeted radiotherapy using 111In-labeled somatostatin derivatives (12,28) and persuaded by the interesting findings on the therapeutic effects of internalizing antibodies labeled with Auger electron–emitting radionuclides (6), we envisaged the design of new trifunctional conjugates of a truncated analog of somatostatin bearing a function for receptor binding and internalization (TOC): one for the nucleus transfer (NLS; PKKKRKV) and one for the cytotoxic or reporter effect (111In-DOTA) (19). Combining these two targeting strategies, we aimed to achieve a higher accumulation of radioactivity in the cell nuclei and a prolonged retention time of the radioligand in the tumor cells, both conditions being necessary for effective Auger electron DNA cytotoxicity transfer. As the pharmacologic profile of DOTA-TOC–like conjugates is greatly influenced by any structural alterations, we incorporated the NLS in 3 different positions relative to the somatostatin analog sequence, respectively. The first experiment done to test the best assembly of the trifunctional derivatives (1), (2), and (3) was the binding affinity profile to somatostatin sst2 receptors. Because natIn-DOTA-TOC has suitable affinity only for the sstr2 subtype (20), we used HEK cells transfected with sstr2A. Confirming one of our hypotheses, the C-terminal modification of DOTA-TOC (compound 3) showed a significant loss in binding affinity for this receptor, whereas the N-terminal functionalizations (compounds 2 and 3) preserved the pharmacologic integrity. This result was also confirmed in the internalization studies, with 111In-(1) displaying significantly lower internalization rates in both AR4-2J and HEK-sstr2A cell lines in comparison with 111In-DOTA-TOC. On the other hand, the 111In-labeled derivatives (2) and (3) revealed increased specific accumulation in both cell lines. Nevertheless, 111In-(3) proved to be the best design, not only because of the binding affinity but also because it shows the highest internalization rates (at 4 h, 2-fold internalization rate when compared with the parent compound in both cell lines studied). Confirming the premise that the NLS has to be located in the cytoplasm to drive the cargo to the nucleus (18), 111In-(4) has no uptake and no binding in both the rat (AR4-2J) and the human (HEK-sstr2A) cell lines.
In addition to the high specific cellular uptake, 111In-(3) reveals also a low externalization rate in HEK-sstr2A cells. Although this derivative seems to reach a plateau after 4 h of externalization, with >70% of the 2-h internalized radiopeptide retained intracellularly, the 111In-DOTA-TOC retained in the cell is only 40% and continues to decrease gradually. The gap between the 2 externalization rates becomes even clearer when the cellular retention is expressed as a percentage of the added radioligand.
The most explicit confirmation of the hypothesis on which the design of these NLS-somatostatin derivatives was based comes from the nuclei isolation experiment. The fast nuclear targeting is supported by the 11.2% uptake of 111In-(3) in the nuclei in comparison with only 0.7% for 111In-DOTA-TOC at just 1 h after incubation at 37°C. Both compounds show a gradual decrease in the cellular nuclear accumulation from 1 to 24 h, with the ratio of this uptake between the 2 conjugates reaching a maximum at 4 h (82 times higher nuclear uptake for 111In-(3)).
The higher internalization rate, the prolonged cellular retention and the significantly higher nuclear uptake of 111In-(3) in comparison with the parent compound are proofs that the principle of 2-step targeting may work in practice. It was previously shown that some radiolabeled somatostatin analogs translocate to the nucleus after internalization (29,30), usually after longer incubation times. Our results support this theory, but they also show that the addition of a NLS moiety dramatically increases the rate of nuclear targeting, enhancing the efficacy of a potential Auger electron-emitter cytotoxic effect. Although both the complete mechanisms of endocytosis of somatostatin analogs (31) and of translocation through the nuclear pores of NLS-cargo conjugates (32) still remain to be clarified, at this point we can only assume that a part of the internalized radioligand escapes the lysosomal degradation (33). The use of strongly fluorescing lanthanides such as Eu3+ or Tb3+ used as surrogates of In3+ may represent the best proof of nuclear localization of such conjugates (34). Nevertheless, it is not crucial that the radiolabeled conjugate remains intact after endocytosis, being sufficient if only the 111In-DOTA-NLS portion of 111In-(3) arrives in nuclear proximity. A longer retention in the nuclear compartment may necessitate an additional functional group intercalating into the DNA.
CONCLUSION
The research dealing with the molecular design of new targeting agents is rapidly expanding in the field of nuclear medicine. We have designed and characterized in vitro new NLS-conjugated DOTA-somatostatin–based derivatives. The peptides were compared with our clinical gold standard [111In-DOTA-Tyr3]octreotide. These first series of in vitro data demonstrate that the concept of 2-step targeting can work in practice if the conjugates have a suitable design. Future experiments will show the extent to which this approach may improve the cytotoxic effect of such conjugates. This type of strategy could be of real interest particularly for the treatment of disseminated tumor cells, using Auger electron–emitting radionuclides. Or it can be used as a neoadjuvant therapy in combination with β-emitting targeted radiotherapy in neoplasia having both large tumors and small malignancies.
Moreover, this model can be extended also to other ligands of G-protein–coupled receptors or to other cytotoxic moieties, thus further improving the targeting strategies.
Acknowledgments
We acknowledge the financial support from the Swiss National Science Foundation (project 3100A0-100390), European Molecular Imaging Laboratories, and COST (European Cooperation in the Field of Scientific and Technical Research) B12 (Action B12: “Radiotracers for the in vivo assessment of biologic functions”).
Footnotes
Received Jun. 16, 2005; revision accepted Aug. 11, 2005.
For correspondence or reprints contact: Helmut R. Maecke, PhD, Division of Radiological Chemistry, Department of Radiology, University Hospital Basel, Petersgraben 4, CH-4031 Basel, Switzerland.
E-mail: hmaecke{at}uhbs.ch
REFERENCES
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Experimental Radionuclide Therapy of HER2-Expressing Xenografts Using Two-Step Targeting Nuclisome Particles
- Epidermal Growth Factor Receptor Inhibition Modulates the Nuclear Localization and Cytotoxicity of the Auger Electron Emitting Radiopharmaceutical 111In-DTPA Human Epidermal Growth Factor
- 111In-Labeled Trastuzumab (Herceptin) Modified with Nuclear Localization Sequences (NLS): An Auger Electron-Emitting Radiotherapeutic Agent for HER2/neu-Amplified Breast Cancer
- Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors
- Auger Electrons: Lethal, Low Energy, and Coming Soon to a Tumor Cell Nucleus Near You