


Atheromas are very prevalent in humans at autopsy. Most of the lesions are found in the aorta and in the carotid, iliac, and coronary arteries. Histologic studies have demonstrated that atheromas go through a cycle of lipid deposition, inflammatory cell infiltration, increased vasa vasorum, plaque disruption—either by erosion (sudden loss of endothelial cells leading to exposure of underlying thrombogenic collagen) or rupture (loss of integrity of the cap of the plaque by weakening at the shoulders of the lesion or sudden increase in pressure within the plaque due to hemorrhage, exposing thrombogenic contents)—platelet deposition, thrombus formation, and reendothelialization, followed by quiescence (Fig. 1). This cycle is repeated multiple times for each lesion, leading to the conclusion that most plaque disruptions are clinically silent (1). This concept is supported by the observation in autopsy studies, as well as during angioscopy of patients with acute coronary syndromes, that patients with an acute thrombus have multiple vulnerable lesions (2–4).
There are 2 potential clinical consequences of thrombus formation on plaque. The first is that emboli arising from the plaque thrombus occlude a critical vessel downstream. The second is that the thrombus occludes a major portion of the vessel lumen at the site of the lesion. Lesions in the aorta are associated with embolism, lesions in the coronaries are associated with occlusion, and lesions in the carotids are associated with both. Because plaques are common and patients can survive for many years with flow-limiting lesions (when the underlying atheroma is stable, such as in chronic stable angina), only a small subset of all atheromas requires immediate treatment. These are the lesions most likely to cause a clinical event.
Libby (5) suggests “… most myocardial infarctions result not from critical blockages but from lesions that produce stenoses that do not limit flow. For example, in individuals who have undergone coronary arteriography in the months preceding myocardial infarction, the culprit lesion most often shows less than 50% stenosis. In a compilation of 4 such serial angiographic studies, only approximately 15% of acute myocardial infarctions arose from lesions with degrees of stenosis greater than 60% on an antecedent angiogram (6,7).”
Atheromas causing a clinical event are often called vulnerable plaque, a term coined by Muller in 1985 (8) in an article describing the circadian variation in the frequency of myocardial infarction (a 3-fold higher incidence of myocardial infarction in the morning [peak at 9 am] than in the evening [trough at 11 pm]). Although vulnerable plaques are a small fraction of the total burden of fibroatheroma (9), these lesions are numerous in patients with clinical events. Histologically, the broad category of vulnerable plaques is characterized by a fibrous cap < 65 μm thick (10); a lipid core occupying at least 50% of the plaque volume; numerous lipid-filled macrophages near the necrotic core and in the cap; abundant lymphocytes in the adventitia (11); and other inflammatory cells, such as mast cells, in the lesion. Histologic reconstruction of lesions that were found at autopsy demonstrated that 70% of lesions had ruptured and healed without causing a clinical event (9). However, compared with calcified fibroatheroma, thin-capped lipid-filled lesions with intense inflammation have a higher likelihood of rupture leading to a major acute coronary event. Recent studies suggest, however, that the composition of atheromas leading to acute events in young patients is different from that seen in patients in their 70s and 80s. In the latter group, the majority of culprit atheromas are calcified, contain limited amounts of lipid and relatively few inflammatory cells, and often cause clinical events through erosion rather than rupture. These age-related differences have a major impact on the potential approaches available to detect patients at high risk of a clinical event.
Although ultrasound (12), CT (13,14), and MRI (15) can identify atheromas on the basis of degree of stenosis, intima-to-media thickness ratio, presence and extent of calcium, or appearance of lipid content, these technologies cannot see a key component of the lesion: inflammation. 18F-FDG PET, on the other hand, can readily identify the metabolic activity of macrophages and other hypermetabolic cells in inflammatory lesions (16). Localization of 18F-FDG in atheromas has been documented for the carotid arteries (17), aorta (18), and other major vessels (19) of humans.
On pages 1245–1250 of this issue of The Journal of Nuclear Medicine, Ogawa and colleagues describe the results of 18F-FDG imaging in rabbits with hereditable hypercholesterolemia (20). The Watanabe rabbit (WR) possesses a heritable trait of hyperlipidemia, with an arterial system lacking functional low-density lipoprotein receptors. Aortic atherosclerosis develops spontaneously by the age of 5 mo (21). A few weeks after birth, fatty streaks develop in the aortae of young WRs; macrophages (i.e., lipid-laden foam cells) are the most prevalent cell type in these streaks (22,23). As young WRs grow older, fatty streaks develop in the intramyocardial and carotid arteries (24). WR atherosclerosis predominates in the arch of the aorta, with diffuse thickening of the vascular wall. Descending thoracic atherosclerosis in WRs varies from a patchwork of atheromatous plaques to more diffuse disease.
Ogawa et al. (20) correlated 18F-FDG uptake determined on ex vivo well-counting studies of 6-mm segments of the aorta with histologic quantitation of the number of macrophages based on the macrophage-specific stain, RAM-11. The investigators found an r value of 0.81 between macrophage number and 18F-FDG uptake.
There is a cautionary note in their results, however. The investigators compared uptake in the hyperlipidemic WRs with that in control animals. 18F-FDG localization was almost 3-fold greater in hyperlipidemic vessels than in control vessels. The in vivo images demonstrated a modest degree of 18F-FDG uptake, which was difficult to identify in images. The clinical challenge is not identifying the patient with atheroma (which can be done through blood lipid measurement and assays of inflammatory factors) but recognizing specific lesions likely to cause clinical events. To fulfill this role, the imaging agent must have a high concentration only in these critical lesions, with a minimal concentration in other atheromas. 18F-FDG may concentrate in too broad a range of lesions to provide unambiguous clinical data. Other markers, such as 99mTc-labeled annexin V, a marker of apoptosis (25), have also been suggested for these studies (26). The pathophysiologic basis for using annexin is the presence of large numbers of apoptotic cells in unstable lesions. At this point it is not clear whether 18F-FDG or annexin (27) or perhaps some other marker will provide the specific localization required to make the technique clinically useful.
Regardless of which tracer proves most useful, however, the relationship between tracer localization and macrophage activity in the plaque is likely to be a key to identifying lesions likely to cause clinical events.
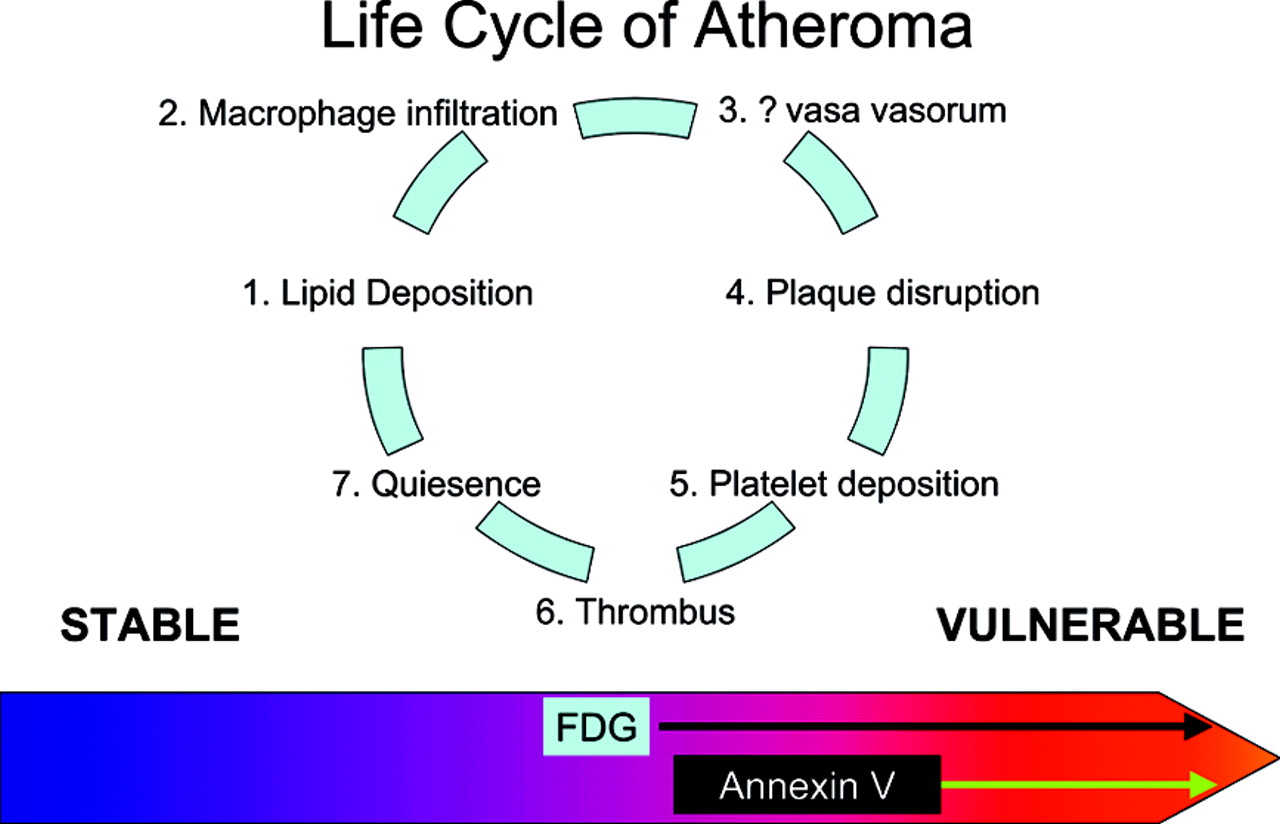
Diagram depicting the life cycle of an atheroma. The lesion goes through several cycles of endothelial injury, lipid deposition, macrophage attraction, increased vasa vasorum, and quiescence. The plaque may progress to the point where its cap is disrupted, leading to platelet deposition and thrombus formation. Most of these episodes are clinically silent. The lesion heals, undergoes reendothelialization, and may remain quiescent for decades. The major goal is to identify lesions likely to cause a clinical event in the near term. Several tracers can accomplish this task. The leading contenders appear to be 18F-FDG (metabolic activity of the inflammatory cells) and annexin V (apoptosis of cells in the lesion). 18F-FDG, however, may identify a broader range of lesions than does annexin and, as such, may be less helpful as a marker identifying patients for intervention and intense local therapy.
Footnotes
Received May 21, 2004; revision accepted May 27, 2004.
For correspondence or reprints contact: H. William Strauss, MD, Nuclear Medicine Service, Memorial Sloan Kettering Cancer Center, Room S212, 1275 York Ave., New York, NY 10021.
E-mail: straussH{at}mskcc.org
REFERENCES
In this issue





{kind=link}
Jump to section
Related Articles
Cited By...
- Imaging Atherosclerotic Plaque Inflammation by Fluorodeoxyglucose With Positron Emission Tomography: Ready for Prime Time?
- Multimodality Imaging of Carotid Artery Plaques: 18F-Fluoro-2-Deoxyglucose Positron Emission Tomography, Computed Tomography, and Magnetic Resonance Imaging
- Detection and Treatment of Vulnerable Plaques and Vulnerable Patients: Novel Approaches to Prevention of Coronary Events