


Abstract
Hypertrophic cardiomyopathy (HCM) is a genetic disease caused by mutations in genes encoding sarcomeric proteins. However, other genetic and possibly also environmental factors modify the phenotypic expression of left ventricular (LV) hypertrophy. The present study investigated whether cardiac adrenergic activity affects the severity of LV hypertrophy in genetically identical patients with HCM. Methods: The study population consisted of 21 patients with HCM caused by the Asp175Asn substitution of the α-tropomyosin gene (TPM1-Asp175Asn) and 9 healthy volunteers. LV mass and segmental wall thickness were measured with MRI. Presynaptic cardiac adrenergic activity was measured with 123I-metaiodobenzylguanidine (MIBG) SPECT. Global and segmental washouts of 123I-MIBG were calculated. Results: Global myocardial 123I-MIBG washout was faster in patients with TPM1-Asp175Asn than in healthy volunteers (50% ± 9% vs. 37% ± 8%, P = 0.001). In linear regression analysis, global 123I-MIBG washout was associated with the LV mass index and LV maximal wall thickness index in HCM patients (r = 0.512, P = 0.018, and r = 0.478, P = 0.028, respectively). The mean 123I-MIBG washout was higher in LV segments ≥ 15 mm thick than in LV segments < 15 mm thick (56 ± 10 vs. 49% ± 10%, P = 0.002). Conclusion: In patients with HCM sharing the same causal gene defect, the degree of LV hypertrophy is related to 123I-MIBG washout, suggesting that cardiac adrenergic activity modifies phenotypic expression in HCM.
Hypertrophic cardiomyopathy (HCM) is a genetic disorder caused by various mutations in 11 genes encoding sarcomeric proteins (1,2). However, these mutations only partially account for the variability in left ventricular (LV) hypertrophy (LVH) and other phenotypic features of the disease. It has been suggested that in patients with HCM, the final phenotype, including LVH, is the product of the causal mutation in genes encoding sarcomeric proteins, modifier genes, and environmental factors (1). The I/D polymorphism of the angiotensin-I converting enzyme gene has been shown to modify LVH in HCM, but this polymorphism accounts for only <15% of the variability in hypertrophy (3). Other genetic and environmental factors responsible for the variability in LVH in HCM remain largely unknown.
HCM is characterized not only by LVH but also by diastolic dysfunction, myocardial ischemia, and increased risk of sudden cardiac death (4,5). Many of these features could be explained by exaggerated myocardial adrenergic activity. First, norepinephrine induces myocardial cell growth, disarray, and scarring (6–13). Second, α-adrenergic coronary vasoconstriction can induce myocardial ischemia (14). Third, norepinephrine increases the rate of spontaneous depolarization of myocardial cells, which may predispose to ventricular arrhythmias. Fourth, most patients with HCM have a high global LV ejection fraction, indicating increased contractility, which might be due to accelerated cardiac adrenergic activity. Finally, β-adrenergic blocking agents have been shown to be efficient in controlling symptoms in HCM, and in children with HCM, β-blocker therapy probably improves survival (4,5,15). Although there is evidence that cardiac adrenergic activity plays a central role in HCM, few previous clinical studies have investigated cardiac adrenergic drive in patients with this disease (16–24), and to our knowledge, there are no previous studies on cardiac adrenergic activity in patients with a genetically identical cause for HCM.
Radioiodinated metaiodobenzylguanidine (123I-MIBG) provides an opportunity to noninvasively explore cardiac adrenergic neuronal function in vivo. 123I-MIBG shares the same neuronal transport and storage mechanisms with norepinephrine (25). Several clinical conditions associated with increased cardiac adrenergic tone have been shown to be characterized by accelerated 123I-MIBG washout (26,27). Also, in HCM patients, 123I-MIBG washout has been found to be accelerated (16,17).
To determine the relationship between hypertrophy and cardiac adrenergic activity in HCM, we combined 2 tomographic imaging methods, 123I-MIBG SPECT and cine MRI, in patients with HCM caused by the Asp175Asn substitution of the α-tropomyosin gene (TPM1-Asp175Asn). First, we studied the association between global cardiac adrenergic activity and LV mass, and second, we investigated the relationship between segmental cardiac adrenergic activity and segmental LV wall thickness.
MATERIALS AND METHODS
Subject Selection
The Kuopio University Hospital region in eastern Finland covers a population of approximately 250,000 inhabitants. All patients with suspected or confirmed HCM in this area are referred to the Kuopio University Hospital, Division of Cardiology, Department of Medicine, for diagnosis and treatment. Previously, all unrelated patients from this area with suspected or confirmed HCM were examined at Kuopio University Hospital by the same cardiologist. Altogether, 36 consecutive unrelated patients aged 16 y or more and fulfilling the criteria for definite HCM were identified (28). In the index patients, the clinical diagnosis of HCM was based on the demonstration of an LV maximal wall thickness of at least 15 mm on 2-dimensional echocardiography in the absence of other causes for ventricular hypertrophy, such as arterial hypertension (28). Clinical diagnosis of HCM in adult relatives of the probands was based on the diagnostic criteria by McKenna et al. (29). Briefly, relatives with an LV maximal wall thickness of at least 13 mm on 2-dimensional echocardiography in the absence of other causes for ventricular hypertrophy were classified as having HCM, as well as those patients with pathologic Q waves on the electrocardiogram (ECG) (29).
Variants in genes encoding sarcomeric proteins were screened using the polymerase chain reaction–single-strand conformation polymorphism method and direct sequencing, as previously described (28,30).
Of the 36 index patients with HCM in the Kuopio area, 4 had the TPM1-Asp175Asn, accounting for 11% of all HCM cases in this area (28). Of 37 relatives of these 4 index patients, 22 were found to have the TPM1-Asp175Asn mutation. In addition, 1 family from western Finland having 4 family members and the identical TPM1 gene mutation was included in the study. Of all these subjects, 6 were not willing to participate, and in 3 cases 123I-MIBG imaging was not feasible. Finally, the population of the present study consisted of 21 adult subjects from 5 families with the TPM1-Asp175Asn. All these 5 kindreds with the TPM1-Asp175Asn (also the family from western Finland) carried the same repeat-sequence allele of the HTMαCA marker (28), suggesting that the Asp175Asn mutation may have arisen from a common ancestor in these families. This ancestor, however, must be distant, since 3–5 generations of each family have been studied and, so far, no common ancestor or kinship has been found (28).
All HCM patients underwent an interview, physical examination, 12-lead ECG recording, and echocardiography. The physical examination included measurement of height and weight. Blood pressure was measured twice on the right arm after 10 min of rest. Echocardiographic evaluation was performed with a Sonos 1000 or 5500 scanner (Hewlett-Packard, Palo Alto, CA) with a 2.5-MHz transducer. All measurements were performed according to the standards of the American Society of Echocardiography (31). Coronary angiography was performed in 19 of 21 HCM patients to exclude coronary artery disease.
Healthy Volunteers
Nine healthy volunteers were studied (4 men and 5 women; mean age ± SD, 46 ± 6 y). None of the healthy volunteers was taking any medication. Blood pressure was measured after 10 min of rest. The 12-lead ECG was obtained and a blood sample for measurement of fasting glucose was drawn to exclude diabetes.
The study protocol was approved by the Kuopio University Hospital Ethics Committee, and all subjects gave written informed consent.
Assessment of LVH with MRI
In patients with HCM, cine MRI was performed to calculate LV mass and segmental wall thickness. MRI was performed with a 1.5-T clinical MRI scanner (Magnetom Vision; Siemens Medical Systems Inc., Erlangen, Germany). A phased-array body coil was used as a receiver. All images were obtained during repeated breath-holds and were ECG gated. Localizer images were obtained to define the imaging planes to the LV short-axis orientation. Imaging was done at the short-axis orientation from the mitral valve insertion to the apex without gap (6–11 slices; slice thickness, 10 mm; voxel size, 2.0 × 1.2 × 10.0 mm).
At each short-axis level, surface areas lined by endocardium and epicardium were manually traced, excluding papillary muscles and trabeculations. Total LV end-diastolic volume was achieved by summing the end-diastolic endocardial volumes at each section, which were calculated by multiplying each traced end-diastolic endocardial area by the slice thickness (10 mm). End-diastolic epicardial volume was calculated in an identical manner by tracing the epicardial area at each section. To evaluate myocardial volume, we subtracted endocardial volume from epicardial volume. Myocardial mass was calculated by multiplying myocardial volume by a myocardial density of 1.05 g/mL (32).
The LV maximal wall thickness at diastole of all the imaged short-axis slices from mitral valve insertion to apex was noted. The thickness of the true apex was determined from the long-axis images. Because LV mass and LV maximal wall thickness are dependent on body size, the LV mass index and LV maximal wall thickness index values were used in analysis. LV mass and LV maximal wall thickness indices were calculated by dividing the LV mass by body surface area. Body surface area was calculated by the following equation: [weight (kg) × height (cm)/3,600]0.5 (33).
Assessment of Cardiac Adrenergic Activity with 123I-MIBG
In HCM patients, 123I-MIBG imaging was performed during the same day as MRI. All imaging was performed with a MultiSPECT 3 gamma camera (Siemens Medical Systems, Inc., Hoffman Estates, IL) equipped with high-resolution collimators. A dose of 200 MBq of 123I-MIBG (specific activity, 26,000 MBq/mmol) (MAP Medical Technologies Oy, Helsinki, Finland) was injected into the antecubital vein during rest. The energy window of 123I was set at 162 keV (150–174 keV). The first acquisition was started 15 min after injection, and the second acquisition was started 4 h later using the same image settings. An anterior planar study was acquired using a 5-min acquisition time and a 128 × 128 matrix size. During SPECT acquisition, 3 detectors (3 × 120°) acquired 30 views in 4° steps of 40 s per view with a 64 × 64 matrix size. In the healthy volunteers, imaging was done in a similar way.
To assess the 123I-MIBG uptake differences between HCM patients and healthy volunteers, data of initial and delayed planar imaging were used. By using similarly sized rectangular regions of interest, we were able to normalize mean planar cardiac 123I-MIBG counts by the mean upper mediastinal counts of the respective image, expressed as heart-to-mediastinum (H/M) ratio (34).
123I-MIBG washout between 15 min and 4 h was calculated as follows: (A − B)/A × 100, where A is 123I-MIBG uptake in the initial image and B is 123I-MIBG uptake in the delayed image. No physical decay correction of 123I was applied.
To assess the association between LVH and global and segmental 123I-MIBG washout, data from SPECT imaging were used. The SPECT technique overcomes the limitations related to planar imaging, such as superposition of noncardiac structures, and enables the assessment of regional radioligand uptake. The raw data of 123I-MIBG SPECT were reconstructed using a Butterworth-filtered (order, 6; cutoff frequency, 0.5/cm) backprojection technique. Global myocardial washout was calculated as a mean value of 4 different segmental washouts (anterior, septal, lateral, and inferior myocardial regions). The utmost apical and basal slices of each region were excluded to ensure the myocardial origin of detected activities.
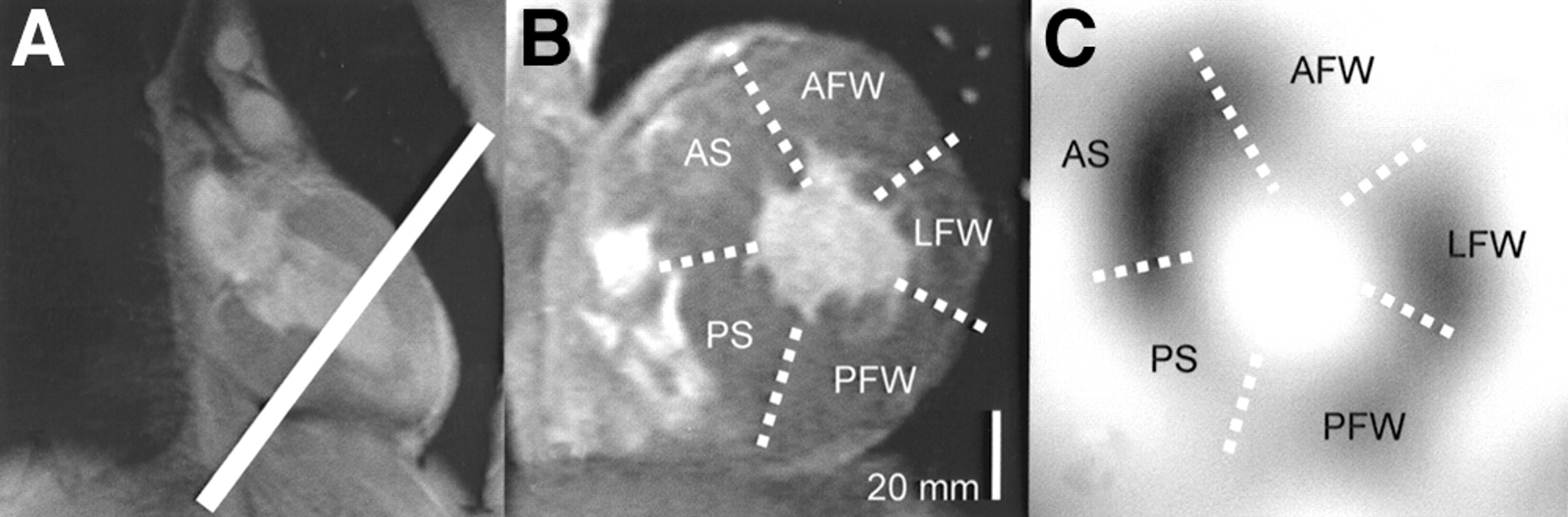
Segmental analysis was performed at the midventricular level at short-axis orientation. The most middle short-axis tomogram in both MRI and SPECT was selected to represent the mid ventricular level. The LV short axis was divided into 2 septal (anterior and posterior) and 3 free-wall (anterior, lateral, and posterior) segments. End-diastolic LV wall thickness by MRI and 123I-MIBG washout were measured in each segment (Fig. 1).

Images show how segmental wall thickness and segmental 123I-MIBG washout were measured. (A) Long-axis MR image of left ventricle. White line indicates midventricular level. (B) Short-axis MR image at midventricular level. White dotted lines indicate borders of 5 short-axis segments, maximal thickness of which was measured. (C) Short-axis reconstruction of 123I-MIBG SPECT image. White dotted lines indicate borders of 5 left-ventricular short-axis segments defined in similar way as in short-axis MRI. 123I-MIBG washout in these segments was measured. AFW = anterior free wall; AS = anterior septum; LFW = lateral free wall; PFW = posterior free wall; PS = posterior septum.
Statistical Analysis
All data are expressed as the mean value ± SD. A t test for independent samples was used to test the differences in 123I-MIBG activity between HCM patients and healthy volunteers, and 123I-MIBG washout between LV segments ≥ 15 mm and < 15 mm thick. Because of skewed distribution, LV mass index was analyzed after logarithmic transformation. Linear regression analysis was used to study the association between global 123I-MIBG washout and the LV mass index and between global 123I-MIBG washout and the LV maximal wall thickness index. P ≤ 0.05 was considered statistically significant. Statistical analyses were performed with a statistical software package (SPSS Win 9.0.; SPSS Inc., Chicago, IL).
RESULTS
Clinical Findings
The clinical findings for HCM patients are summarized in Table 1. Two of 21 patients did not fulfill the echocardiographic or electrocardiographic criteria for HCM. In addition, 2 of the 19 HCM patients with a hypertrophied left ventricle had previously diagnosed arterial hypertension and 2 had somewhat high blood pressure in the present study. Because patients with previously diagnosed arterial hypertension were now normotensive, and because, in those with high blood pressure in the present study, the elevation was only mild, we considered those 4 patients to also fulfill the diagnostic criteria for HCM. For practical reasons, all subjects with the TPM1-Asp175Asn substitution, and the aforementioned 2 subjects who did not meet the diagnostic criteria for HCM, are called HCM patients in the following comparisons.
Clinical Characteristics of Subjects with Asp175Asn Substitution of α-Tropomyosin Gene
On 2-dimensional echocardiography, the mean maximal end-diastolic thickness of the septum was 19 ± 6 mm (range, 8–29 mm). LV end-diastolic and end-systolic dimensions as well as LV ejection fraction were within normal limits in all HCM patients. The most common cardiac symptoms were palpitations and dyspnea. One subject had undergone a myotomy–myectomy operation. One third of the subjects were taking cardiac medication, most often β-blockers. Most subjects had abnormal ECG findings. Coronary angiography was performed on 19 of 21 HCM patients. None of the patients had coronary artery stenosis exceeding 50% of the luminal diameter, but 2 patients had <50% stenosis in 1 of the main branches.
Healthy volunteers had normal resting ECG, normal blood pressure (systolic blood pressure, 127 ± 13 mm Hg; diastolic blood pressure, 85 ± 9 mm Hg), and normal fasting blood glucose (4.6 ± 0.3 mmol/L).
LV Mass and LV Maximal Wall Thickness in MRI
In MRI, the mean LV mass in HCM patients was 155 ± 60 g (range, 80–313 g). The mean LV maximal wall thickness in MRI was 19.8 ± 5.2 mm (range, 10–31 mm). In 3 of the HCM patients, LV maximal wall thickness was <13 mm in the MRI study.
Cardiac Adrenergic Activity in HCM Patients and Healthy Volunteers
At 15 min, the H/M uptake ratio was similar between patients with HCM and healthy volunteers (1.70 ± 0.15 vs. 1.67 ± 0.09, P = 0.689). At 4 h, the H/M ratio was lower in HCM patients than in healthy volunteers (1.57 ± 0.16 vs. 1.77 ± 0.20, P = 0.008). Thus, because of the higher turnover of 123I-MIBG, global washout was faster in HCM patients than in healthy volunteers (50% ± 9% vs. 37% ± 8%, P = 0.001) (Fig. 2).

Global 123I-MIBG washout in patients with HCM caused by Asp175Asn mutation of α-tropomyosin gene and in healthy volunteers.
Association Between Cardiac Adrenergic Activity and LVH
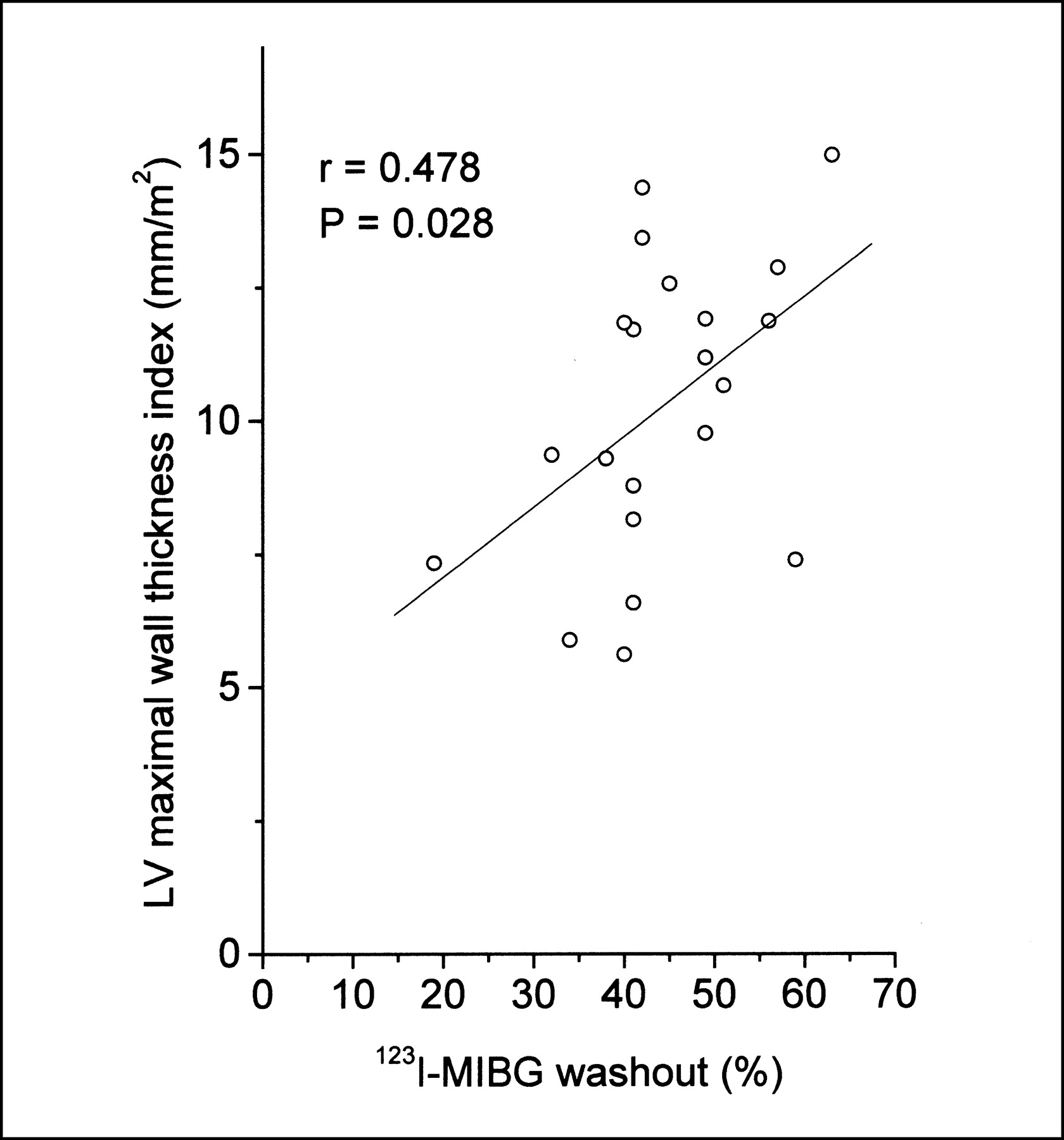
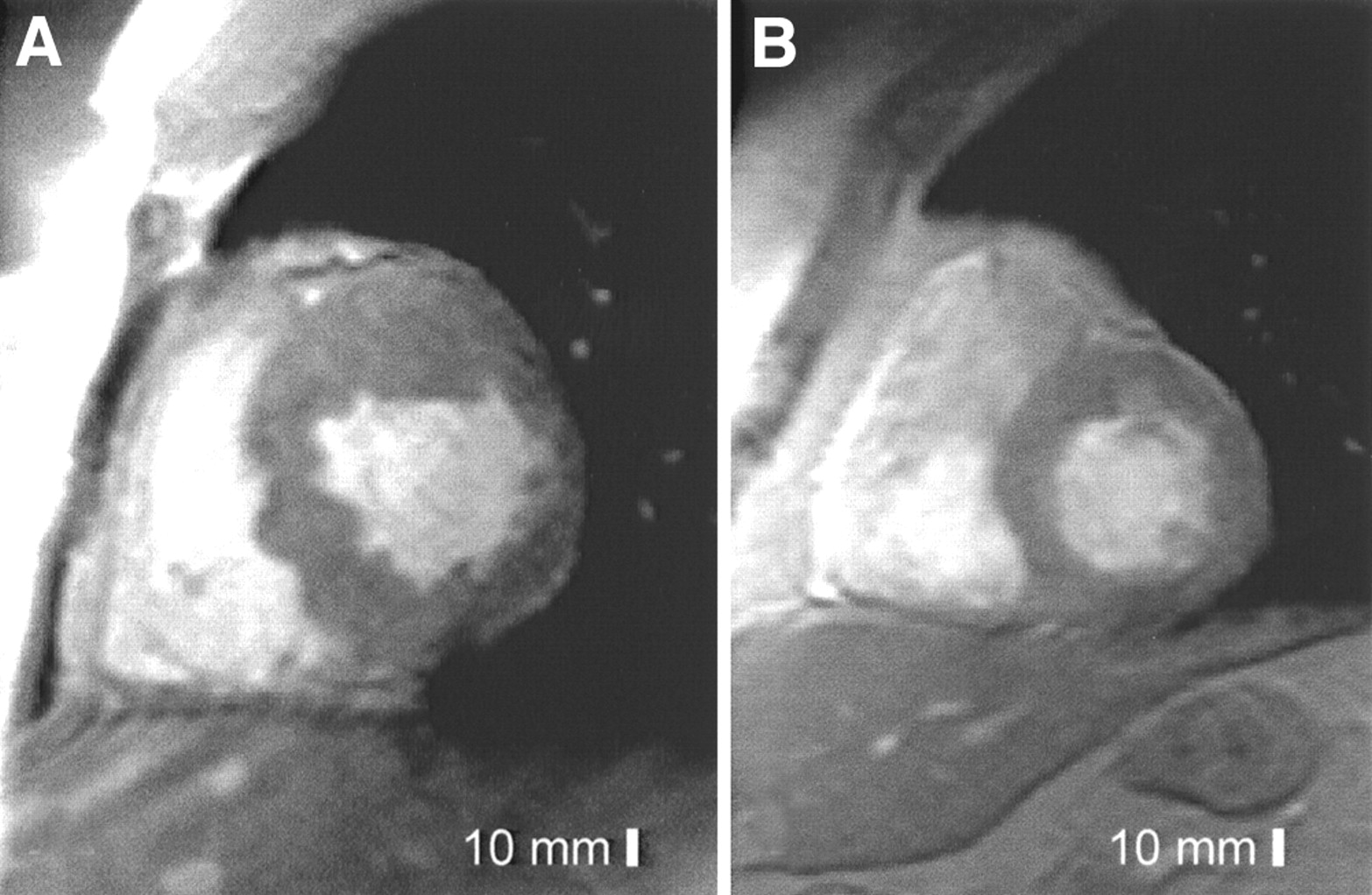
In linear regression analysis, global 123I-MIBG washout was associated with the LV mass index and LV maximal wall thickness index in HCM patients (r = 0.512, P = 0.018, and r = 0.478, P = 0.028, respectively) (Figs. 3 and 4). Two subjects with the TPM1-Asp175Asn substitution from the same family with a different extent of LVH and MIBG washout are illustrated in Figure 5.

Linear-logarithmic plot of global 123I-MIBG washout and LV mass index in patients with HCM caused by Asp175Asn mutation of α-tropomyosin gene.
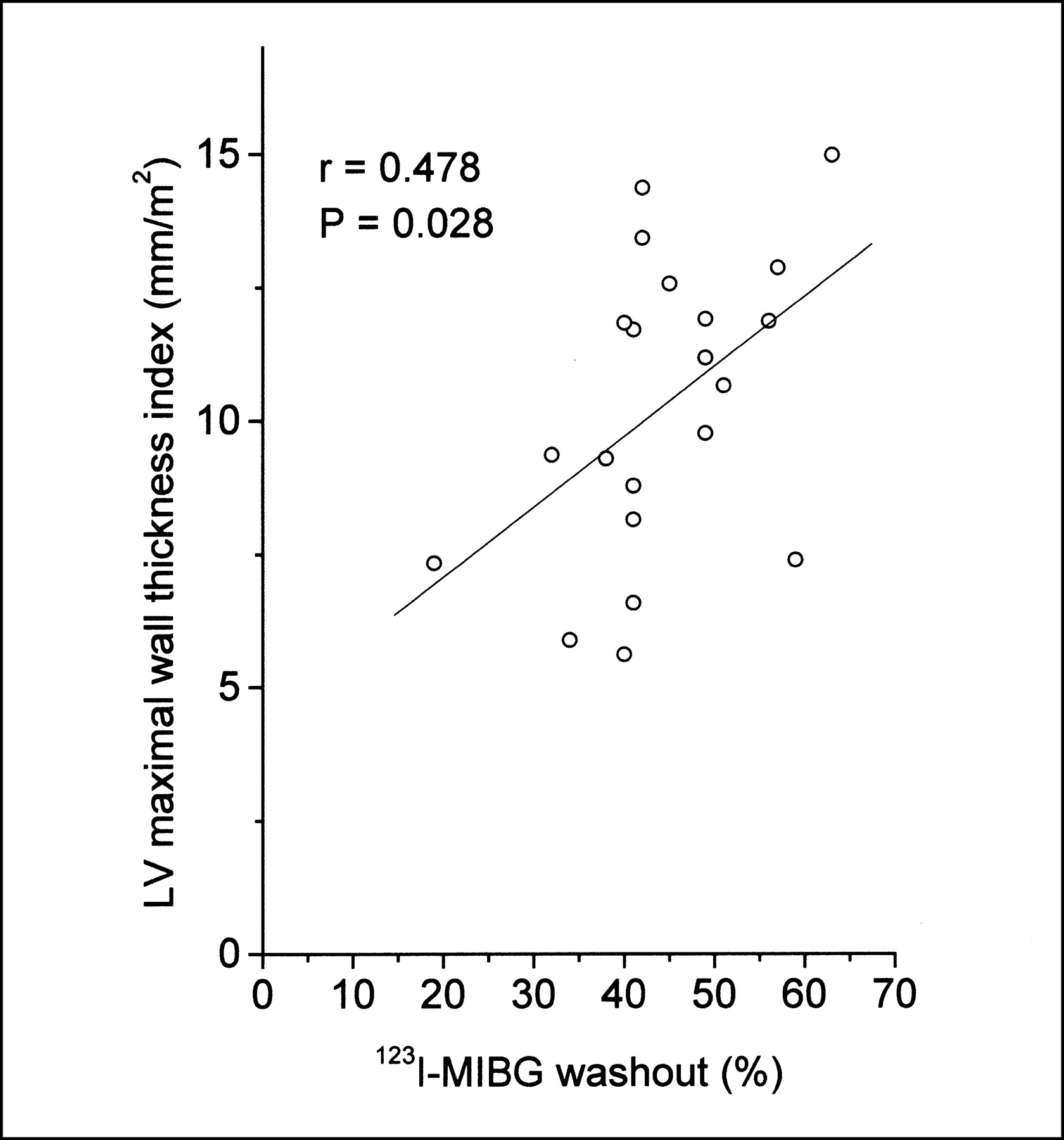
Linear plot of global 123I-MIBG washout and LV maximal wall thickness index and in patients with HCM caused by Asp175Asn mutation of α-tropomyosin gene.

MRI LV short-axis cine images in 2 patients from 1 family with Asp175Asn substitution of TPM1 gene. (A) A 22-y-old man with increased maximal LV thickness (31 mm), increased LV mass index (164 g/m2), and accelerated 123I-MIBG washout (67%). (B) A 48-y-old uncle of previous patient. Maximal LV wall thickness, LV mass index, and 123I-MIBG washout were normal (11 mm, 50 g/m2, and 47%, respectively).
At the segmental level, the mean wall thickness in LV segments ≥ 15 mm thick (n = 26) was 18.8 ± 3.5 mm and the mean wall thickness in LV segments < 15 mm thick (n = 79) was 9.9 ± 2.6 mm. The mean 123I-MIBG washout was higher in LV segments ≥ 15 mm thick than in LV segments < 15 mm thick (56% ± 10% vs. 49% ± 10%, P = 0.002).
DISCUSSION
Many clinical features of HCM and previous studies (18–21) suggest accelerated cardiac adrenergic activity in this disease. However, to our knowledge, no studies comparing LVH and cardiac adrenergic activity in genetically homogeneous HCM patients have been performed. In the present study, 123I-MIBG washout was faster in HCM patients with the TPM1-Asp175Asn substitution than in healthy volunteers, indicating accelerated cardiac adrenergic activity in HCM. Moreover, in patients with HCM, global 123I-MIBG washout correlated with the LV mass and LV maximal wall thickness, suggesting that enhanced cardiac adrenergic activity is one factor explaining the variability of LVH in HCM patients with the TPM1-Asp175Asn substitution.
We found that 123I-MIBG washout was higher in thick than thin segments of the left ventricle. Previous studies have shown somewhat variable results for the association between LVH and adrenergic activity at the regional level. Li et al. (21) found a lowered ratio of 18F-6-fluorodopamine to perfusion in hypertrophied LV regions compared with nonhypertrophied LV regions in HCM patients or compared with that of the septum of healthy volunteers (21). In contrast, Lefroy et al. found no difference in β-adrenoceptor density in the most hypertrophied LV regions compared with the least hypertrophied regions in HCM patients (18). Schäfers et al. found globally altered but regionally similarly distributed 11C-hydroxyephedrine (a catecholamine analog) accumulation between HCM patients and healthy volunteers (20).
Previous work, as well as our study, indicates increased presynaptic cardiac adrenergic activity and downregulated postsynaptic activity in patients with HCM (16–21). Our study has, however, several methodologic advantages compared with previous studies. First, in our study, HCM patients represented a wide spectrum of hypertrophy, since the study group included patients with no hypertrophy, patients with mild LVH, and patients with marked LVH. Second, LVH in this study was measured with MRI, which allows a more comprehensive evaluation of LVH than does echocardiography (35). MRI has also been shown to be more accurate and reproducible in the assessment of LV mass than is echocardiography (36). Third, regional analysis was performed by combining 2 tomographic imaging modalities, 123I-MIBG SPECT and MRI, thus enabling the assessment of regional radioligand activity and hypertrophy segment by segment.
All previous studies on cardiac adrenergic activity in HCM have been performed in patients who have not been genetically characterized (18–21,24). Although many of these studies have shown that adrenergic activity is accelerated in patients with HCM, and some studies have even shown that sympathetic activity is associated with the extent of LVH, these findings could be explained by different genetic defects causing HCM in these patients. Different defects in sarcomeric genes induce LVH to a variable extent (1), and variable myocardial sympathetic activity might also be related to different causal mutations. In the present study, however, all patients had the same gene defect of the TPM-1 gene. Consequently, the association between accelerated adrenergic activity and LVH shown by the present study cannot be attributed to variable causal mutations.
Previous studies have shown that norepinephrine induces hypertrophy of cultured rat myocytes (7,9) and that prolonged infusion of subhypertensive doses of norepinephrine in dogs induces an increase in the mass of the myocardium and the thickness of the LV wall (6,11). Moreover, a recent study on dopamine β-hydroxylase knockout mice has shown that endogenous norepinephrine and epinephrine are required for the induction of in vivo pressure-overload LVH, indicating a central role of cardiac adrenergic activity in myocardial hypertrophy (12,37). In contrast, to our knowledge, no studies have indicated that LVH induces accelerated adrenergic activity. Recently, a hypothesis has been presented that the primary abnormality in the pathogenesis of HCM is decreased contractility of cardiac myocytes that causes increased cell stress and expression of stress-responsive trophic factors, including cardiac adrenergic activation, which in turn induces LVH (38). Accordingly, the findings of the present study suggest that accelerated myocardial adrenergic activity is an important modifier of LVH in HCM.
Recent studies have indicated that the initial phase in the development of myocardial hypertrophy involves the formation of cardiac para- or autocrine factors such as norepinephrine or angiotensin II, the receptors of which are coupled to G proteins (13). A recent study indicated that transgenic mice lacking both Gαq and Gα11 in cardiomyocytes showed complete lack of LVH in response to pressure overload (13). Possibly, targeting this signaling process might prevent myocardial hypertrophy not only in pressure overload hypertrophy but also in HCM.
Sudden cardiac death (SCD) is a major problem in HCM, often affecting young, asymptomatic subjects. Recent studies have shown that the degree of LVH is an important prognostic factor for SCD (39). The risk of SCD increases from 0 per 1,000 person-years in HCM patients with an LV maximal wall thickness ≤ 15 mm to 18.2 per 1,000 person-years in HCM patients with an LV maximal wall thickness of 30 mm or more (39). In HCM, LVH is variable not only in different causal genotypes but also in patients with identical causal gene defects (1). Factors modifying hypertrophy independent of causal mutations are incompletely understood. The notion that cardiac adrenergic activity is associated with LVH in genetically identical patients is an important novel finding, raising interesting new questions, such as what affects sympathetic activity in HCM, and could LVH, and subsequently also SCD, be prevented by modifying sympathetic activity in this disease.
CONCLUSION
The results of the present study indicate that in genetically homogeneous subjects with HCM, cardiac adrenergic activity influences the phenotypic expression of LVH. Further studies are needed to confirm the significance of sympathetic activity as a modifier of LVH in different genotypes of HCM and to determine whether β-blocker therapy, or other possible therapies modulating sympathetic activity or its signaling process, can prevent the development of LVH and improve survival in HCM patients.
Acknowledgments
This study was financially supported by the Kuopio University Hospital EVO-fund (5063502), the Finnish Nuclear Medicine Association, and the Astra Research Fund.
Footnotes
Received Jul. 8, 2002; revision accepted Oct. 8, 2002.
For correspondence or reprints contact: Petri Sipola, MD, Department of Clinical Radiology, Kuopio University Hospital, Puijonlaaksontie 2, FIN-70210 Kuopio, Finland.
E-mail: petri.sipola{at}kuh.fi




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}