


Abstract
Physiologic barriers to the delivery of macromolecules to solid tumors are a major obstacle to the clinical success of radioimmunotherapy (RIT). Only a small fraction of the injected dose of the radiolabeled monoclonal antibody (mAb) localizes at the tumor site. This situation worsens as the tumor burden increases. It is hypothesized that improvements to RIT of adenocarcinoma can be realized by inclusion of vasoactive agents, in particular agents able to increase the vascular permeability of tumor capillaries. In these studies, a response-selective peptide agonist of human C5a, GCGYSFKPMPLaR (AP), was used to transiently increase tumor vascular permeability in an effort to improve RIT of solid tumors. Methods: Athymic mice xenografted with human colorectal adenocarcinoma LS174T were treated intravenously with low doses (9.25 MBq) of 131I-labeled mAb B72.3 in combination with various intravenous doses of AP. The progression of the disease or the loss of >20% body weight was taken as the endpoint. Biodistribution and tumor uptake kinetics were studied in the same tumor-antibody system. Results: The uptake of 125I-B72.3 in LS174T xenografts increased in a dose-dependent manner with an apparent maximal effect between 3 and 6 h after intravenous administration of AP. Augmenting the dose of 9.25 MBq 131I-B72.3 with a single administration of 0.1 mg AP delayed tumor growth nearly 2-fold; the tumor quadrupling time (Tq) was 14.2 ± 3.3 d for 131I-B72.3 alone versus 26.0 ± 3.6 d for 131I-B72.3 plus 0.1 mg AP (P < 0.001). An additional dose of 0.1 mg AP 24 h after 131I-B72.3 further improved the therapeutic outcome (Tq = 48.5 ± 7.9 d; P < 0.001) and resulted in several cases of tumor regression. Conclusion: The inclusion of agonist peptides of human C5a in the RIT scheme results in improved tumor responses without any manifest side effects.
Monoclonal antibodies (mAbs) have significantly improved the targeted delivery of various therapeutic agents and radioisotopes to tumors. However, gains in selectivity are strongly counteracted by problems related to the heterogeneity of the structure and physiology of solid tumors. These physiologic barriers to the delivery of macromolecules are a major impediment to the clinical success of radioimmunotherapy (RIT) of adenocarcinoma. Only a small fraction of the injected dose of the radiolabeled mAb localizes at the tumor site. This situation worsens as the tumor burden increases (1–4). Even in experimental cancer models in mice, in which most small tumors are treated successfully with a single dose of radioimmunoconjugate, the therapeutic response to RIT abates as the tumor size increases (5). The accretion of radiolabeled mAbs and, consequently, radiation doses deposited in tumors rely on the blood flow in and around tumor masses, the tumor vascular volume, and the vascular permeability (VP) of tumor vessels to macromolecules (6–8). All of these factors are particularly impaired in large tumors, which have disordered vasculature and lack draining lymphatic systems. Methods to transiently change the tumor VP have been suggested as a means to increase the delivery of mAbs to tumors. Increases in tumor uptake, some significant, are reported, when the mAb delivery is aided by external beam irradiation (9), hyperthermia (10), and systemically administered vasoactive agents, such as angiotensin II (11), interleukin-2 (12), tumor necrosis factor-α (13), and, most recently, αvβ3 antagonist cyclo-RGD-d-phenylalanine-1-aminocyclohexane carboxylic acid (14). However, to date, the improved therapeutic outcome resulting from the modulation of the VP during RIT has not been documented.
The human anaphylatoxin C5a is a multifunctional phlogistic molecule that is generated as a cleavage product of the fifth component of complement during serum complement activation. One characteristic of the C5a-mediated activity is the increase of the VP to facilitate the recruitment of circulating neutrophils, monocytes, eosinophils, and basophils to sites of tissue injury or infection (15,16). C5a-derived small agonist peptides, which maintain this biologic property, are expected to improve the delivery of radiolabeled mAb to tumors. On the basis of the structural analysis of a panel of conformationally restricted peptides with homology to the C-terminal end of human C5a, a biologically preferred peptide conformation was defined. Peptides with optimal biologic activities have residues 65–69 in a twisted, helix-like conformation, whereas residues 70–71 exist in an extended backbone conformation, and residues 71/72–74 exist in a B-turn of either type II or type V for spasmogenic responses and a B-turn of type V for neutrophil responses (17,18). One analog with a Pro-for-Asp substitution at position 71 and a d-Ala-for-Gly substitution at position 73, YSFKPMPLaR (C5a65–74, Y65, F67, P69, P71, d-Ala73), a = d-Ala, expressed 15%–20% of the full C5a activity in spasmogenic assays and approximately 5% in polymorphonuclear (PMN) assays. YSFKPMPLaR was also shown to be stable in the presence of mouse and human serum carboxypeptidases (19). In this study a response-selective peptide agonist of human C5a, GCGYSFKPMPLaR (AP), was used to transiently increase tumor the VP in an effort to improve RIT of solid tumors. AP is an analog of YSFKPMPLaR, a conformationally biased, response-selective agonist of human C5a that produces marked increases in the VP in the skin of guinea pigs and hypotensive effects in rats (19,20). Structure-activity studies have shown that the conformational features expressed by YSFKPMPLaR and its analog AP are more amenable to interacting with and activating C5a receptors expressed on cells responsible for hemodynamic activities than for C5a receptors expressed on inflammatory PMN cells (17–20). This report shows unequivocally that RIT combined with the VP-enhancing C5a agonist peptide GCGYSFKPMPLaR improves the outcome of RIT.
MATERIALS AND METHODS
Peptides and mAbs
AP was synthesized by standard solid-phase methods, purified, and characterized according to previously described procedures (17).
mAb B72.3 was produced by the University of Nebraska Medical Center Monoclonal Antibody Facility and purified from mice ascites by protein-G affinity chromatography. B72.3 recognizes a high-molecular-weight glycoprotein complex designated as tumor-associated glycoprotein 72 (TAG-72) and shows reactivity with >85% of adenocarcinomas with minimal reactivity to normal tissues (21).
Iodination of mAbs
B72.3 was labeled with Na125I (biodistribution) or Na131I (therapy) using the IODO-GEN (1,3,4,6-tetrachloro-3α,6α-diphenylglycoluril; Pierce, Rockford, IL) method. Briefly, to prepare 131I-B72.3 for RIT studies 2 mg B72.3 were reacted with 370 MBq Na131I in the presence of 0.1 mg IODO-GEN as the oxidant. The unbound radioisotope was removed on a desalting column (Econo-Pac 10DG; Bio-Rad, Hercules, CA) equilibrated with 0.01 mol/L phosphate buffer, 0.0027 mol/L KCl, 0.137 mol/L NaCl (phosphate-buffered saline [PBS]), pH 7.4, at 25°C. Quality assessment using instant thin-layer chromatography with 20% aqueous methanol as the solvent indicated that >95% of the radioiodine was routinely protein bound. Specific activities of 150–190 MBq 131I per milligram B72.3 and 290–330 MBq 125I per milligram B72.3 were typical. The immunoreactivity of radiolabeled B72.3 was tested using bovine submaxillary gland mucin ([BSM] Sigma, St. Louis, MO) attached to Reacti-Gel beads (Reacti-Gel HW-65F; Pierce). Beads coated with bovine serum albumin (BSA) were used as the negative control. Radiolabeled B72.3 (100,000 counts per minute [cpm]) was incubated for 1 h with 0.1 mL BSM- and BSA-beads at room temperature. The unbound B72.3 was removed by repeated washing with PBS containing 1% BSA and 0.1% Tween 20. The radioactivity in the bead pellet was determined in a γ-counter, and the percentage bound to BSM-coated beads was calculated.
Animal and Tumor Models
Athymic female mice (nu/nu; National Institutes of Health, Bethesda, MD) (average weight, 18 g; age, 4–6 wk) were used. Mice were housed in the University of Nebraska Medical Center Animal Facility, which is fully accredited by the American Association for Accreditation of Laboratory Animal Care. Mice had free access to food and water and were kept on a 12-h light cycle. Potassium iodide-supplemented water was provided 3 d before and for 7 d after any treatment with radioiodinated antibodies. Subcutaneous tumors (20–200 mm3) were produced in these mice approximately 10 d after a right flank injection of 5 × 106 LS174T human colon carcinoma cells in 0.2 mL minimum essential medium ([MEM] GIBCO-BRL, Gaithersburg, MD). The cells were obtained from subconfluent monolayers grown in MEM supplemented with 10% fetal bovine serum.
Immunogenicity of AP
Swiss Albino (n = 6) and C57BL/6 (n = 6) mice were given 3 daily doses (either intradermally or intravenously) of 0.1 mg AP; 2 wk after the last dose, blood was collected through a cardiac puncture and serum was separated and tested for 125I-AP binding. Serum samples were incubated with 300,000 cpm 125I-C5a (specific activity, 740 MBq/mg) for 2 h and tested using either a size-exclusion centrifugation or a 96-well plate binding assay (data not shown). Serum samples were also analyzed by nondenaturing, nonreducing polyacrylamide gel electrophoresis (PAGE). These samples were incubated with 300,000 cpm 125I-C5a (specific activity, 740 MBq/mg) for 12 h at 0°C–4°C. Mouse IgG, IgMκ, and IgMλ were used as negative controls.
Biodistribution of 125I-C5a
To investigate the in vivo fate of AP, the peptide was radioiodinated with no-carrier-added 125I to a specific activity of 0.37 MBq/0.1 mg using IODO-GEN as an oxidant. Mice bearing subcutaneous LS174T xenografts were treated with 0.1-mg doses of 125I-AP (0.37 MBq) administered intravenously in 0.2 mL PBS through a tail vein and euthanized at designated times.
Biodistribution Studies
Mice with LS174T xenografts received an intravenous dose of AP in 0.2 mL 0.1% albumin in PBS through the tail vein. To determine the optimum dosing sequence for AP and B72.3, a time interval between the injection of AP and 125I-B72.3 was varied from 0.5 to 24 h. AP was injected intravenously through a tail vein 0.5, 1, 3, 6, and 24 h before the dose of 125I-B72.3 was given. At the times indicated above after AP, an intravenous dose of 0.37 MBq 125I-B72.3 in 0.2 mL 0.1% albumin in PBS was administered. Control mice were treated with 125I-B72.3 alone. Additional doses of AP were administered at 24 and 48 h after injection of 125I-B72.3 in 2 groups of mice. Seventy-two hours later, mice were euthanized and the necropsy was performed. The weight and radioactive content of blood, lung, heart, spleen, liver, kidney, uterus, muscle, stomach, small intestine, large intestine, skin, and tumor were determined.
To examine the overall effect of AP on the distribution of macromolecules and to access the dose dependence, a nonspecific mouse IgG labeled with 125I was used as a tracer. Athymic mice (without tumors) received a simultaneous injection of AP and 125I-IgG (0.37 MBq per mouse) through the tail vein. Thirty minutes later, the aforementioned tissues were harvested and their radioactive content was determined.
Tumor Therapy Studies
Mice with 10-d-old subcutaneous LS174T xenografts were assigned to 3 therapy groups (9.25 MBq 131I-B72.3 plus 0.1 mg AP, 9.25 MBq 131I-B72.3 plus 2 × 0.1 mg AP, and 9.25 MBq 131I-B72.3 plus 2 × 0.01 mg AP) and 2 control groups (9.25 MBq 131I-B72.3 alone or 0.1 mg AP alone) so that the tumor size distribution was similar in each group. AP in 0.2 mL 0.1% albumin in PBS was administered intravenously, which was followed 3 h later by an intravenous dose of 9.25 MBq 131I-B72.3 in 0.1% albumin in PBS. Control groups received a sham injection of 0.2 mL 0.1% albumin in PBS in place of either AP or 131I-B72.3. In studies with 2 AP doses, the second administration of AP was 24 h after 131I-B72.3 administration. Drinking water was supplemented with 0.04% potassium iodide 3 d before treatment and for 7 d after treatment to protect thyroid function. The tumor size and body weight were measured 3 times a week. Tumor volume ([V] mm3) was calculated as: V = 4/3π · (½ of longer tumor dimension in mm) · (½ of shorter tumor dimension in mm)2. All therapy experiments were terminated when the size of tumor in control groups was 2,000 mm3. Premature euthanasia was also indicated if any ulcer formation was observed or the weight loss was 20% of the initial body weight. Serum collected during the necropsy was analyzed by gel-filtration high-performance liquid chromatography (HPLC) (TSK G2000SW and TSK G3000SW, 0.75 × 30 cm; Toso Haas, Tokyo, Japan; connected in tandem with PBS at a flow rate of 0.5 mL/min) and sodium dodecyl sulfate-PAGE ([SDS-PAGE] 4%–15% acrylamide gradient) to determine the effect of AP on the metabolic fate of 131I-B72.3. The immunoreactivity of all radioactive fractions collected from an HPLC column also was measured using the bead-binding assay described above.
Data Evaluation
Each tumor quadrupling time (Tq) was calculated as the average of the Tq of all LS174T tumors in a given group of mice. Comparison was made using the 2-tailed t test for difference in means.
RESULTS
Dose-Dependence Studies
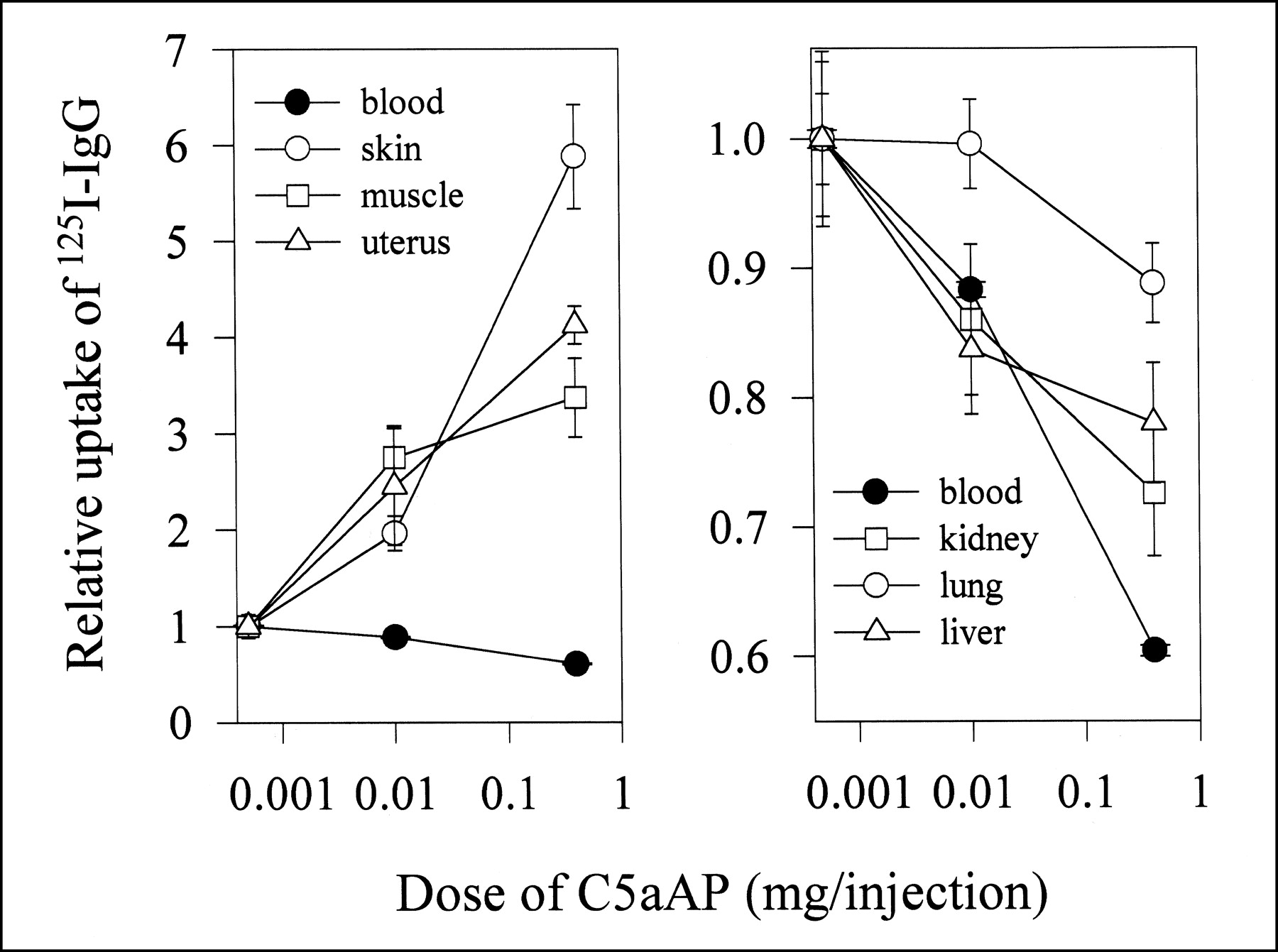
Permeability of mouse tissues to immunoglobulin was evaluated 30 min after a simultaneous intravenous administration of 0.37 MBq 125I-IgG and escalating doses of AP. The egress of 125I-IgG from the intravascular space into the extravascular space depends on the AP dose and is most pronounced in skin, muscle, uterus, and stomach (data for stomach are not shown because data nearly overlap with the uptake in the uterus), but not in other parts of the gastrointestinal tract (Figs. 1A and 1B). Although, the VP increase is dose dependent, this is not a linear relationship (i.e., a 0.4-mg dose of AP does not produce a response 40 times greater than a 0.01-mg dose) (Table 1).

Effects of AP (C5aAP) on uptake of macromolecular tracer in normal tissues of athymic (nu/nu) mice. Dose-dependent changes in uptake of 125I-IgG in various tissues and blood of athymic mice were measured 30 min after intravenous administration of 0.37 MBq 125I-IgG. Mice were given simultaneous injection of 125I-IgG and indicated doses of AP through tail vein. Results are reported as average (n = 4) ± SD. Relative uptake of 125I-IgG is calculated by dividing uptake of 125I-IgG coinjected with AP by uptake of 125I-IgG in absence of AP.
Changes of Tissue Permeability in Response to Treatment with Agonist Peptide GCGYSFKPMPLaR
At the 0.01-mg dose AP, the VP of skin rose by nearly 100%, whereas the 0.4-mg AP dose increased the VP to 850% of the control values. The absolute amount of 125I-IgG localized was 1.5 ± 0.1 percentage injected dose per gram (%ID/g) at 0 mg AP versus 7.8 ± 1.0 %ID/g with 0.4 mg AP. The response to AP appears to saturate at higher doses of AP. For example, gradients of the VP increase are approximately 3 μL blood per gram of skin and 0.6 μL blood per gram of skin per each μg AP at 0.01- and 0.4-mg doses of AP, respectively. The radioactivity accumulated in stomach doubles from 3.1 ± 0.1 %ID/g (0 mg) to 6.9 ± 2.0 %ID/g (0.4 mg). There is also a significant perivascular leakage of radioactivity in uterus, approximately 1.5-fold, reaching 8.9 ± 1.0 %ID/g at this dose of AP. The blood level of 125I-IgG decreases with the increasing dose of AP starting at 44.4 ± 1.1 %ID/g when no AP is administered and plummeting to about one half of this value at 0.4 mg AP. The levels of radioactivity recovered in large and small intestine, liver, spleen, kidney, heart, and lung remain relatively constant with only a slight increase at the highest dose of AP.
Tumor Uptake Changes After AP Administration
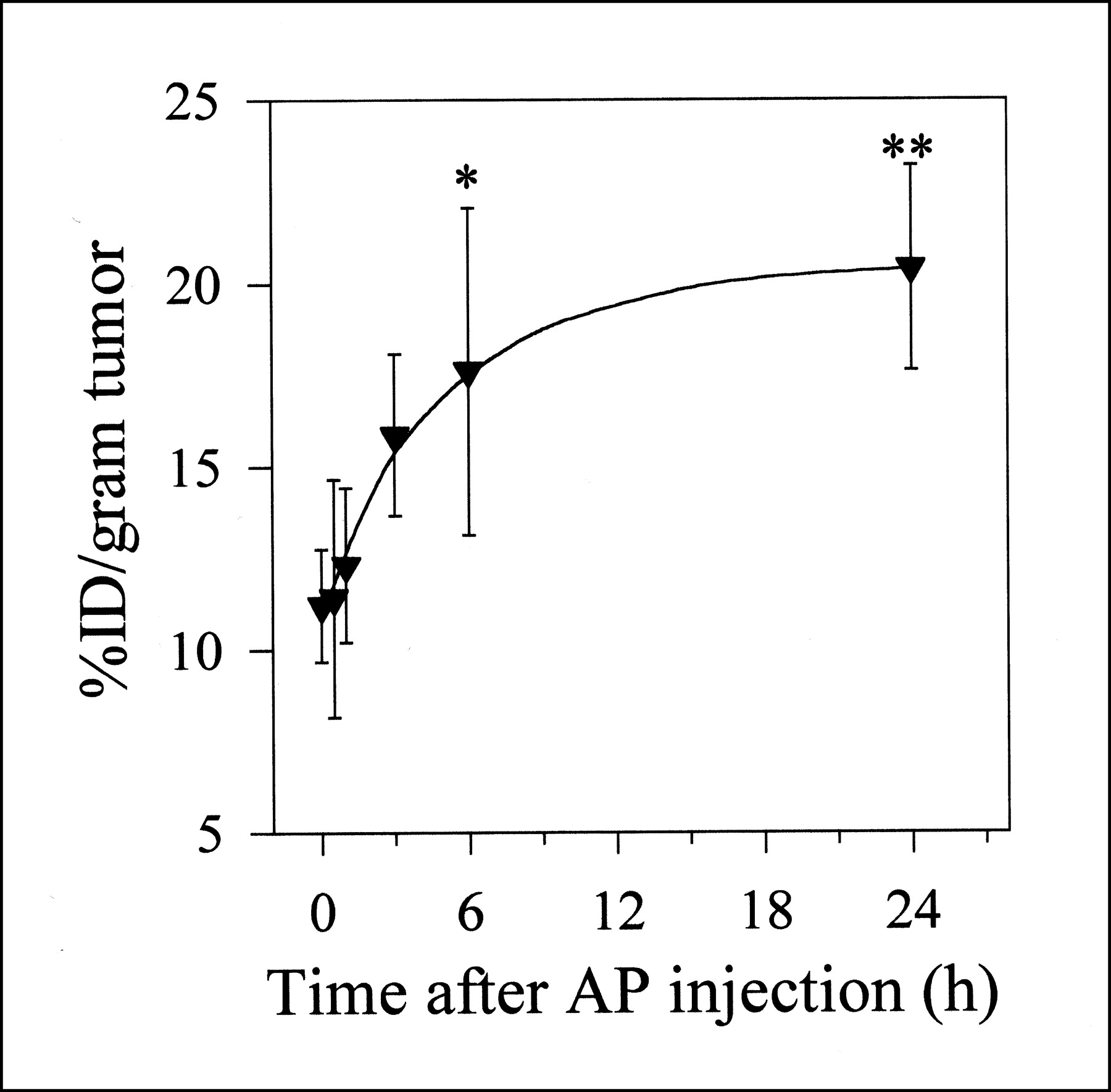
Changes in the 125I-B72.3 uptake in LS174T xenografts as a function of the time interval between the dose of AP and the dose of 125I-B723.3 were measured after 1 intravenous dose of 0.1 mg AP (Fig. 2). 125I-B72.3 was injected either simultaneously with AP (0 h) or at defined intervals (0.5, 1, 3, 6, and 24 h) after AP administration, and the necropsy was performed 72 h after 125I-B72.3 administration. The rate of uptake increase was approximately 6.2%/h during the first 3 h. From 6 h on, the process of extravasation slowed to about 0.34%/h. The best specific uptake in terms of the %ID of 125I-B72.3 per gram of tumor occurred between 3 and 6 h after AP injection. During this period, the distribution of 125I-B72.3 into normal tissues was comparable with that of control values obtained in mice not treated with AP. On the basis of these data, a 3-h time point was selected as an optimum time interval for all therapy studies.

Uptake kinetics of 125I-B72.3 by LS174T xenografts in athymic mice treated intravenously with one 0.1-mg dose of AP at indicated time points before administration of 125I-B72.3. For example, 3 h on x-axis indicates that AP was administered 3 h before dose of radiotracer. Biodistribution was conducted 72 h after injection of 125I-B72.3. Data are expressed as average ± SD (*3-h vs. 6-h uptake, P > 0.2 at 0.05 level of significance; **3-h vs. 24-h uptake, 0.05 > P > 0.02 at 0.05 level of significance).
Effect of Multiple Doses of AP on Uptake of Tumor and Normal Tissues
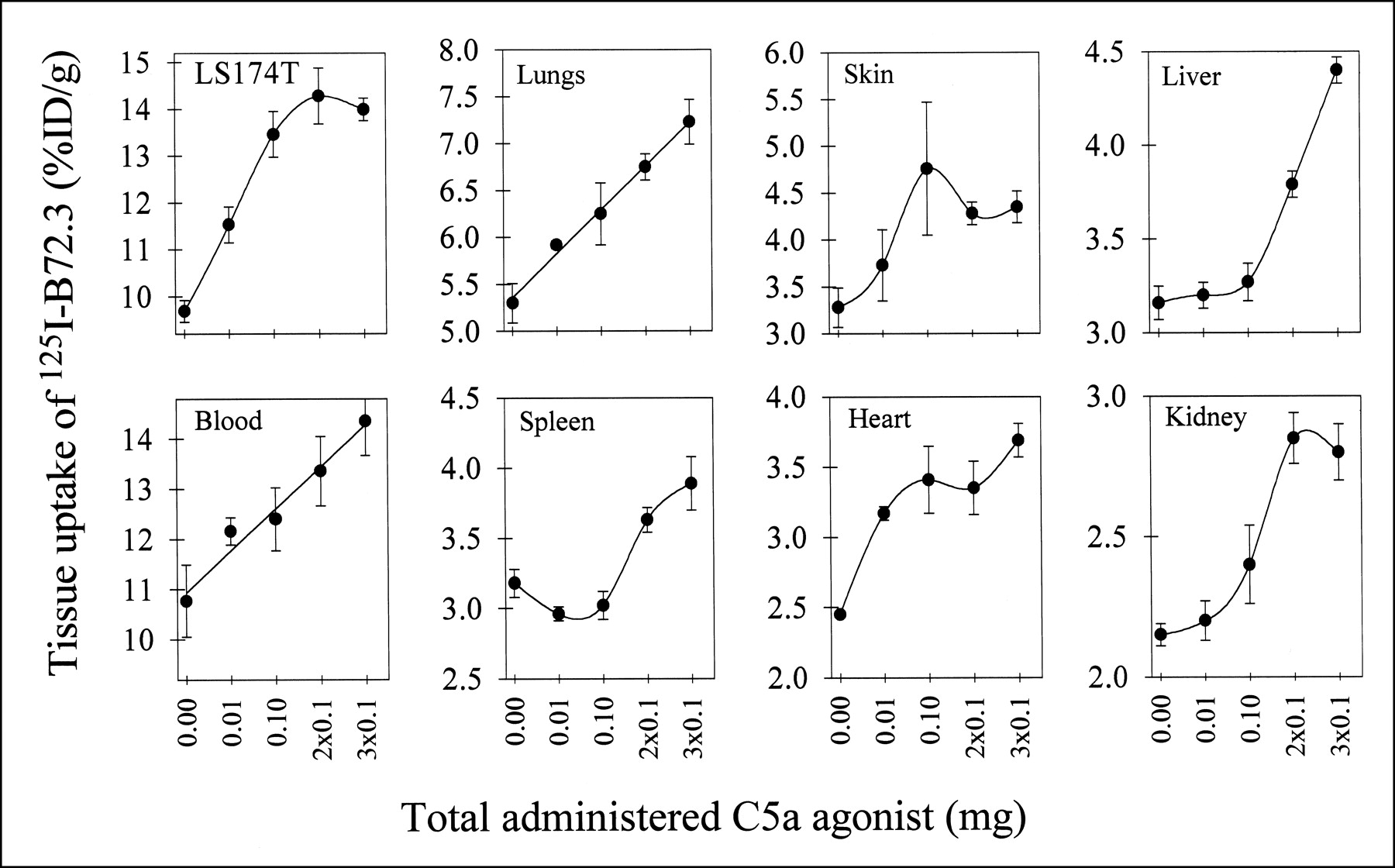
Using the time line established in kinetics studies, AP was injected 3 h before mAb 125I-B72.3 injection, and the necropsy was performed 72 h after administration of 125I-B72.3 (tumor uptake of this and several other radioiodinated murine antibodies is consistently at a peak value at this time point or near this time point). Additional doses of AP were given intravenously in equal fractionated doses every 24 h. Retention of 125I-B72.3 in tumor measured at 72 h increased by approximately 50% with increasing doses of AP, as did the radioactivity levels in blood (i.e., from 10.5 ± 0.7 %ID/g blood with no AP to 14.6 ± 1.3 %ID/g blood at 2 × 0.1 mg AP) (Fig. 3). 125I-B72.3 levels in tumor approach a plateau after three 0.1-mg doses of AP, whereas accumulation of radioactivity continues in several normal tissues (Fig. 3), indicating redistribution and return of the extravasated radioactivity into the bloodstream and normal tissues. This contrasts with necropsy results obtained at 30 min after AP plus 125I-IgG, when the only changes measured are these associated with the VP modifications. At 72 h after 125I-B72.3 administration, the radioactive contents of normal tissues, including skin, parallel blood levels (i.e., there is a gradual increase with escalating doses of AP). However, tumor-to-normal tissue ratios remain similar even at the highest administered dose of AP (3 × 0.1 mg). For example, the tumor-to-liver ratio measured in the absence of AP was 3.5 ± 0.9 compared with 3.2 ± 0.5 after three 0.1-mg doses of AP. Similarly, the tumor-to-kidney ratio stays at approximately 5 in the aforementioned groups of mice. The second dose of AP at 24 h results in further incremental increases of the %ID/g in tumor, and the third dose of AP at 48 h after mAb administration produces a slightly smaller incremental change in %ID/g in tumor from 12.8 %ID/g at 2 × 0.1 mg AP to 13.1 %ID/g at 3 × 0.1 mg AP. On average, the buildup of radioactivity in tumors amounts to approximately 3–4 %ID/g per each 0.1-mg AP. On the basis of these data, the therapy studies were conducted either with 1 dose of AP or with a booster dose 24 h later.

Uptake of 125I-B72.3 in tumor-bearing mice 72 h after injection of radiotracer. 125I-B72.3 was administered 3 h after first dose of AP. Each point is average ± SE of 6 mice.
For as yet unknown reasons, the levels of radioactivity in spleen drop to approximately 50% of control values with 0.01- and 0.1-mg doses of AP. An additional dose of 0.1 mg AP brings the radioactivity to the control baseline. A similar effect is observed in spleen of normal mice (i.e., athymic mice without tumors) treated with 0.01 mg AP and 125I-IgG. Because this is a 72-h biodistribution, the effect cannot be ascribed to neutropenia or a similar event caused by rapid extravasation. Further studies of this phenomenon are in progress.
Immunogenicity of AP
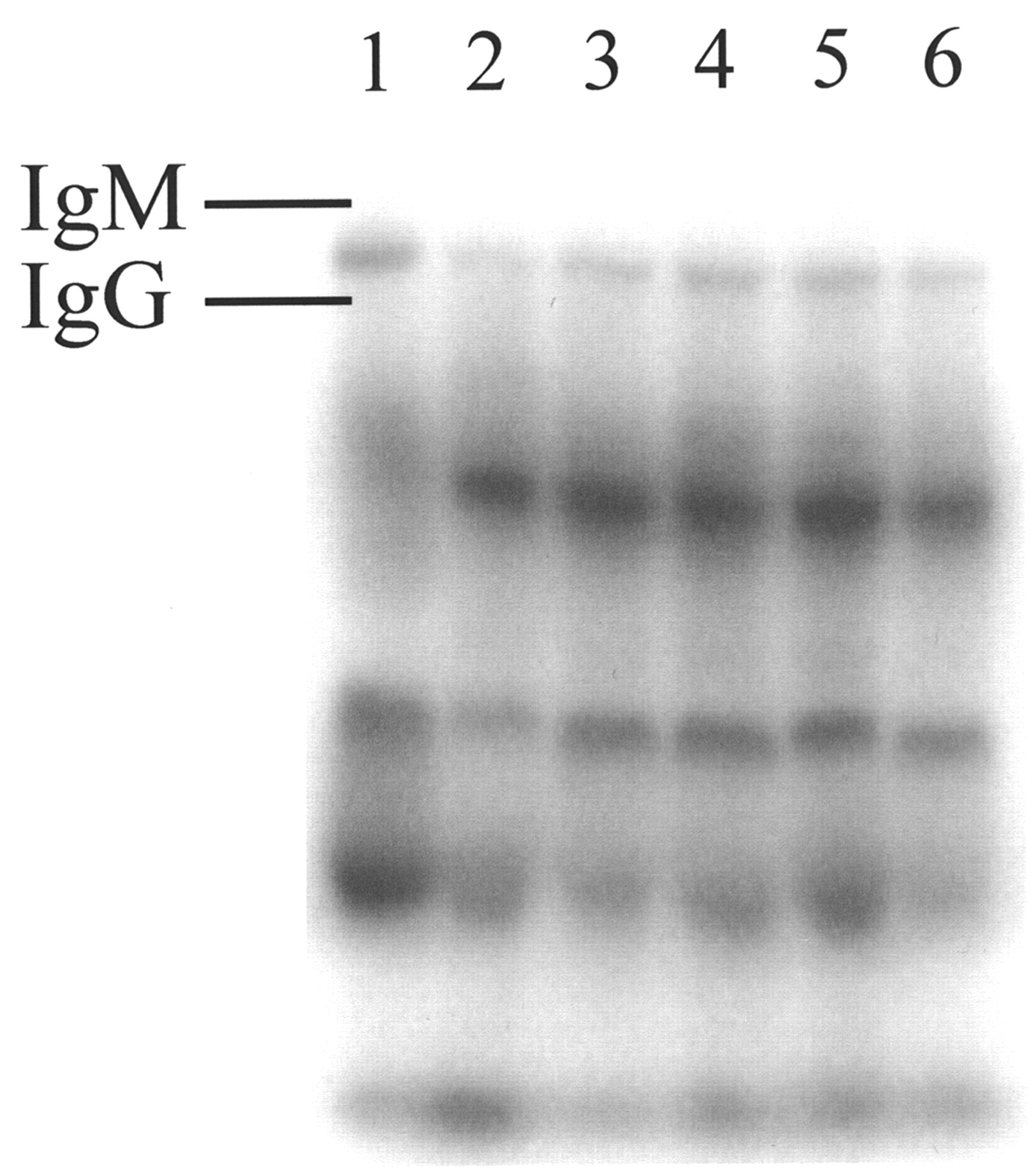
One potential long-term limitation in the use of C5a agonists is a possibility of immunogenic responses when injected into humans. Studies by Tempero et al. (22) have shown that conjugation of an analog of C5a agonists used in our studies, YSFKPMPLaR, to a peptide epitope results in an antibody response to the peptide epitope and to the C5a agonist. Therefore, the effect of multiple doses of AP, administered in normal (i.e., not immunodeficient) mice, on the production of anti-AP response was studied. Mice were given 3 daily doses (either intradermally or intravenously) of 0.1 mg AP; 2 wk after the last dose, blood was collected and tested for 125I-AP binding (Fig. 4). Several components in mouse serum bind the radiolabeled peptide, but no difference was found between serum from AP-immunized (Fig. 4, lanes 3 through 6) and untreated mice (Fig. 4, lanes 1 and 2). There is, however, a good indication that the distribution of AP binding proteins in sera from inbred C57BL/6 mice (Fig. 4, lane 1) differs from that seen in mice of Swiss origin.
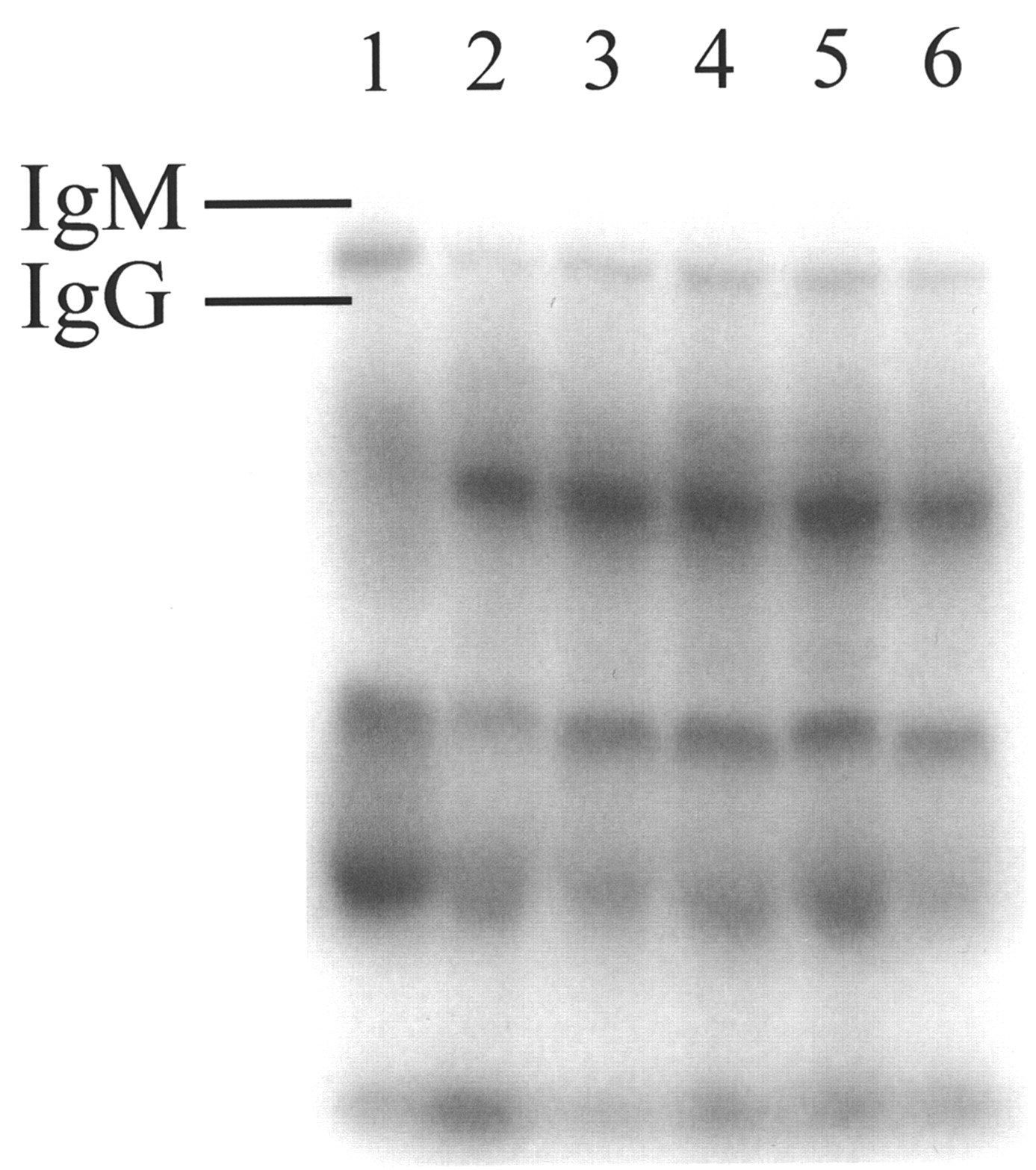
Native PAGE of sera recovered from normal and AP-immunized mice reacted ex vivo with 125I-AP. Mice were given 0.1 mg AP through intradermal or intravenous route once daily for 3 d. Two weeks after last injection, mice were euthanized. Sera collected from mice were incubated with 125I-AP overnight and examined by electrophoresis on native PAGE gel. Lanes: 1, control, C57BL/6 mouse serum; 2, control, Swiss Albino mouse serum; 3, immunized mouse serum, Swiss Albino, intradermal; 4, immunized mouse serum, Swiss Albino, intradermal; 5, immunized mouse serum, Swiss Albino, intravenous; 6, immunized mouse serum, Swiss Albino, intravenous.
Localization of AP in Normal Tissues and Tumors
The delayed effect of AP on biodistribution and tissue perfusion does not correspond to the expected biologic life of low-molecular-weight peptides in vivo. Usually, the blood residence time of radiolabeled peptides is on the order of a few minutes and clearance is rapid from all tissues, with the possible exception of kidneys.
To investigate the in vivo fate of AP, the peptide was radioiodinated with no-carrier-added 125I to a specific activity of 0.37 MBq/0.1 mg and administered intravenously in athymic mice bearing subcutaneous LS174T xenografts. Biodistribution results are shown in Figure 5. The maximum level of 125I-AP in tumors is reached 60 min after injection. Blood clearance curves are typical of substances interacting strongly with blood components. There is a delayed (approximately 60 min after injection) return of radioactivity into the systemic circulation. At this same time point the activity in all normal tissues reaches a second maximum. The time to reach a steady-state concentration is approximately 3 min for all tissues except kidneys (10 min), skin (30 min), and lungs, where only the secondary reabsorption phase is observed. The 125I-AP activity in lungs parallels blood levels at earlier time points but from 30 to 300 min remains relatively constant with an elimination half-life of 19.4 h and a mean residence time of 27 h (uncorrected for 125I decay). Skin-associated AP reaches a maximum after 30 min. The delayed influx of radioactivity into several tissues might indicate an active transport mechanism through the binding and extravasation of PMN leukocyte-125I-AP complexes or the binding of thus activated PMNs to vascular endothelial cells. Both possibilities are being investigated further.
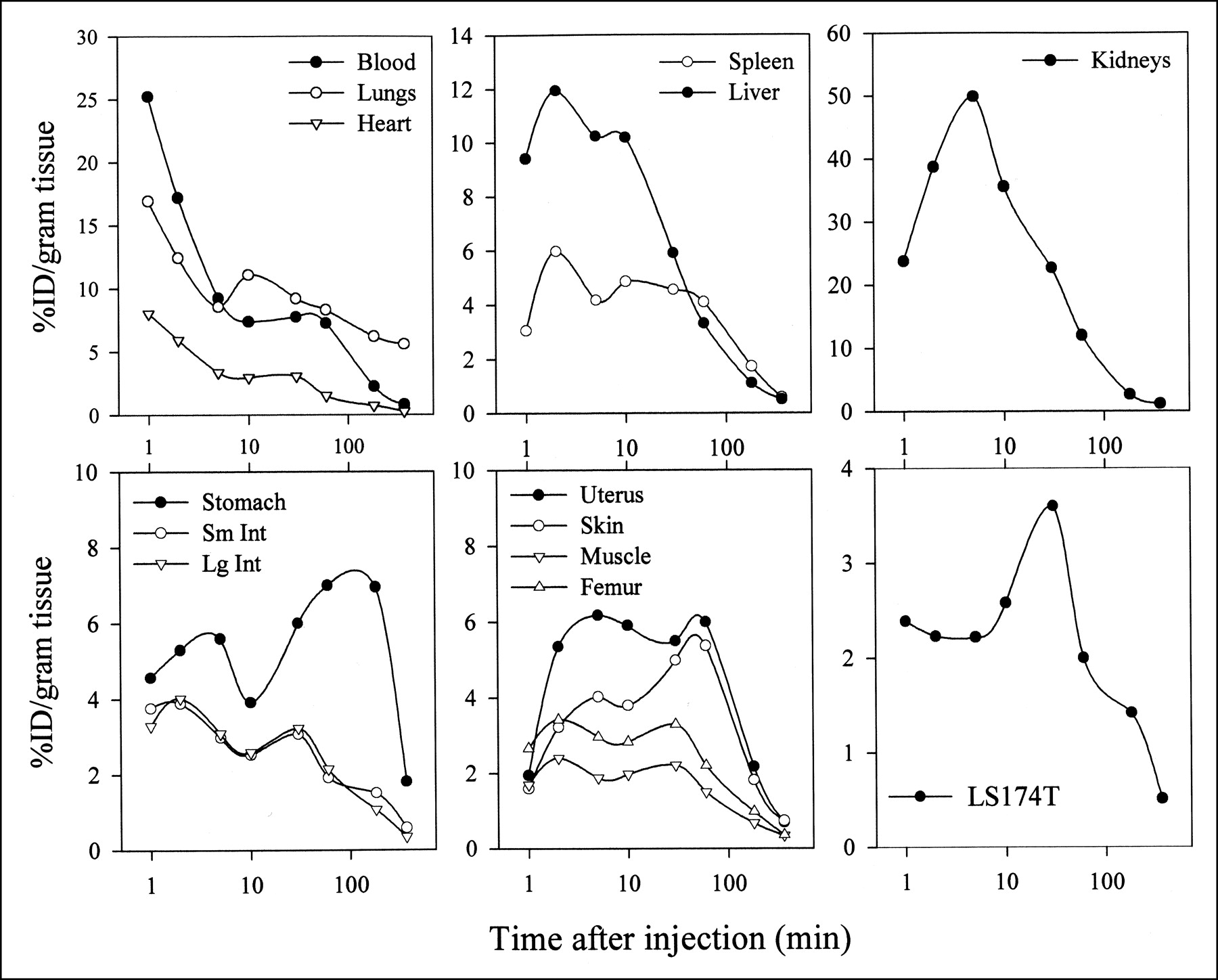
Clearance curves for 125I-AP (0.1 mg) administered intravenously in LS174T-bearing athymic mice. Each symbol represents average ± SD (n = 5); SDs are omitted when they are <5%. Sm Int = small intestine; Lg Int = large intestine.
RIT
Therapy studies were conducted at 3 levels of AP: a single dose of 0.1 mg, 2 doses of 0.1 mg, and 2 doses of 0.01 mg (Tables 2 and 3). The 0.1-mg dose was selected as the optimal dose on the basis of the biodistribution studies. The 0.01-mg doses were included because this was the lowest amount of AP that gave measurable and statistically significant VP changes and tumor uptake increases. The tumor growth rate in the control mice treated only with AP is rapid, with the mean tumor Tq of 7.1 d (Fig. 6A). A similar rapid growth of xenografts is observed in untreated control mice (Fig. 6A, dotted line) and matches our archival data. RIT with 9.25 MBq 131I-B72.3 slows the growth of tumors for approximately 7 d, after which time the growth resumes at approximately the same rate as in the untreated controls (Fig. 6B). The overall Tq in this control is 14.2 d. Even at the lowest dose of AP (i.e., 2 consecutive administrations of 0.01 mg AP 24 h apart), there is a considerable increase in Tq from 7.1 to 34.7 d with the stable disease (i.e., no changes in tumor volume) for about 2 wk. A single dose of 0.1 mg AP is nearly as effective (i.e., the tumor growth delay lasts 2 wk and Tq = 26 d (Fig. 6C). An additional dose of 0.1 mg AP 24 h after 131I-B72.3 administration increases Tq 7 times to 48.5 d. In both groups treated with 2 doses of AP, 4 tumors regressed to 10%–90% of the initial volume. These were excluded in the calculations of Tq.

Relative growth of LS174T colon cancer xenografts after RIT treatment (n = 9). Tumor size changes are expressed relative to tumor volume 10 d after tumor implantation on day of 131I-B72.3 treatment (day 0). (A) AP (0.1 mg) without RIT. Solid lines represent kinetics of tumor growth for each mouse treated with AP; dotted line is mean tumor growth curve in control mice that received no treatment. (B) RIT with 9.25 MBq 131I-B72.3 alone. (C) Combination RIT with 9.25 MBq 131I-B72.3 and 0.1 mg AP.
Summary of LS174T Tumor Growth in Athymic Mice After Combination 131I-B72.3 RIT with 1 Dose of AP
Summary of LS174T Tumor Growth in Athymic Mice After Combination 131I-B72.3 RIT with 2 Doses of AP
Serum samples collected on day 22 of RIT were analyzed by HPLC and SDS-PAGE (Fig. 7) under nonreducing and reducing conditions to examine the metabolic fate of IgG in the presence of AP. On average, the recovered radioactivity amounted to 2.29 ± 0.26 %ID/g and 3.40 ± 0.47 %ID/g of serum (corrected for decay of 131I) in control mice (131I-B72.3 without AP) and treated mice (0.1 mg AP plus 131I-B72.3), respectively. As indicated by HPLC (data not shown) and SDS-PAGE, virtually all 131I recovered in serum was bound to B72.3. The immunoreactivity of serum fractionated on size-exclusion HPLC columns tested in a bead-binding assay is almost identical in different treatment groups (i.e., 48% vs. 51% for 131I-B72.3 alone and 131I-B72.3 plus 0.1 mg AP, respectively, compared with the initial value of 84% on the day of administration).
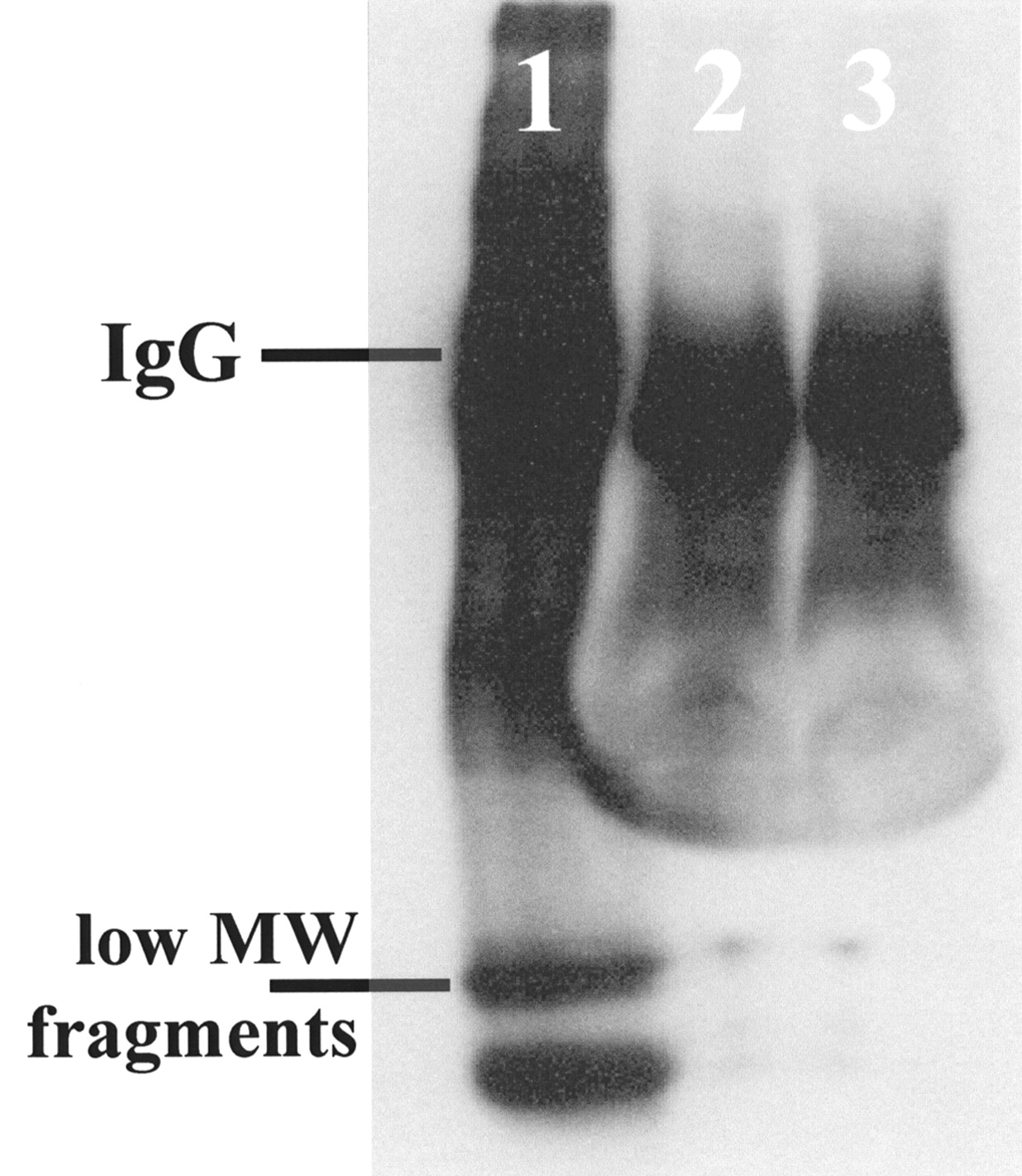
Autoradiogram of 5%–20% gradient SDS-PAGE gel of serum samples indicating no difference in processing of 131I-B72.3 in serum of AP-treated mice compared with 131I-B72.3 alone. Lanes: 1, 131I-B72.3 stored 22 d at 4°C; 2, 131I-B72.3 with AP; 3, 131I-B72.3 alone. All radioactivity recovered in serum 22 d after dosing is in form of 131I-B72.3. HPLC analyses on TSK G2000SW-TSK G3000SW columns confirmed this finding. MW = molecular weight.
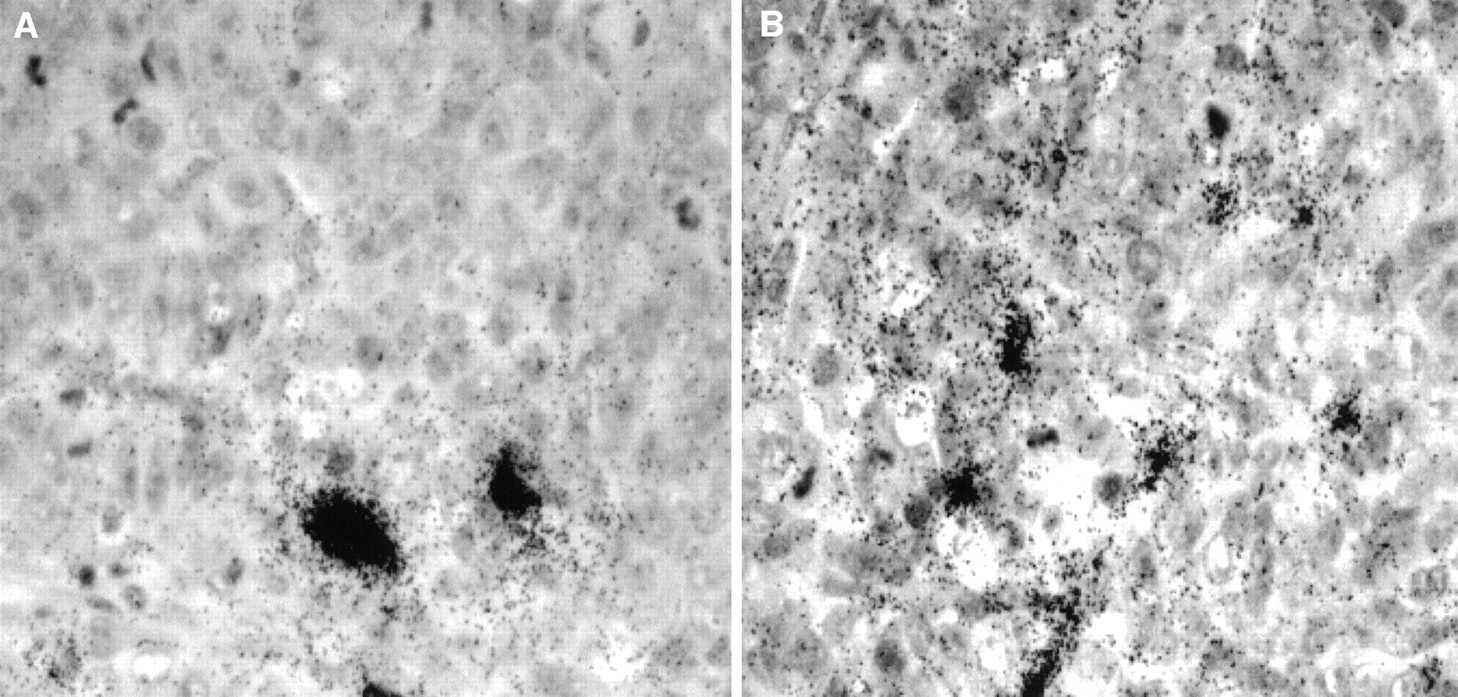
The radioactivity retained in tumors harvested at the same time (22 d) is also similar, 3.95 ± 0.63 %ID/g and 4.70 ± 0.80 %ID/g (corrected for decay) for 131I-B72.3 control mice and the 0.1 mg AP plus 131I-B72.3 treatment group, respectively. However, tumors were considerably smaller in the AP-augmented therapy group, resulting in artificially inflated mean values. The distribution of radioactivity in these tumors from various therapy groups appeared to be more uniform in the AP-treated mice (Fig. 8B). Although on average the values of %ID/g tumor were similar in control and AP-treated mice, autoradiography indicated that in tumor sections from 131I-B72.3 treatment mice a considerable fraction of radioactivity was around blood vessels, whereas the combination of AP plus 131I-B72.3 resulted in the leakage of radioactivity into the extracellular spaces in some regions of the tumor (Fig. 8A).
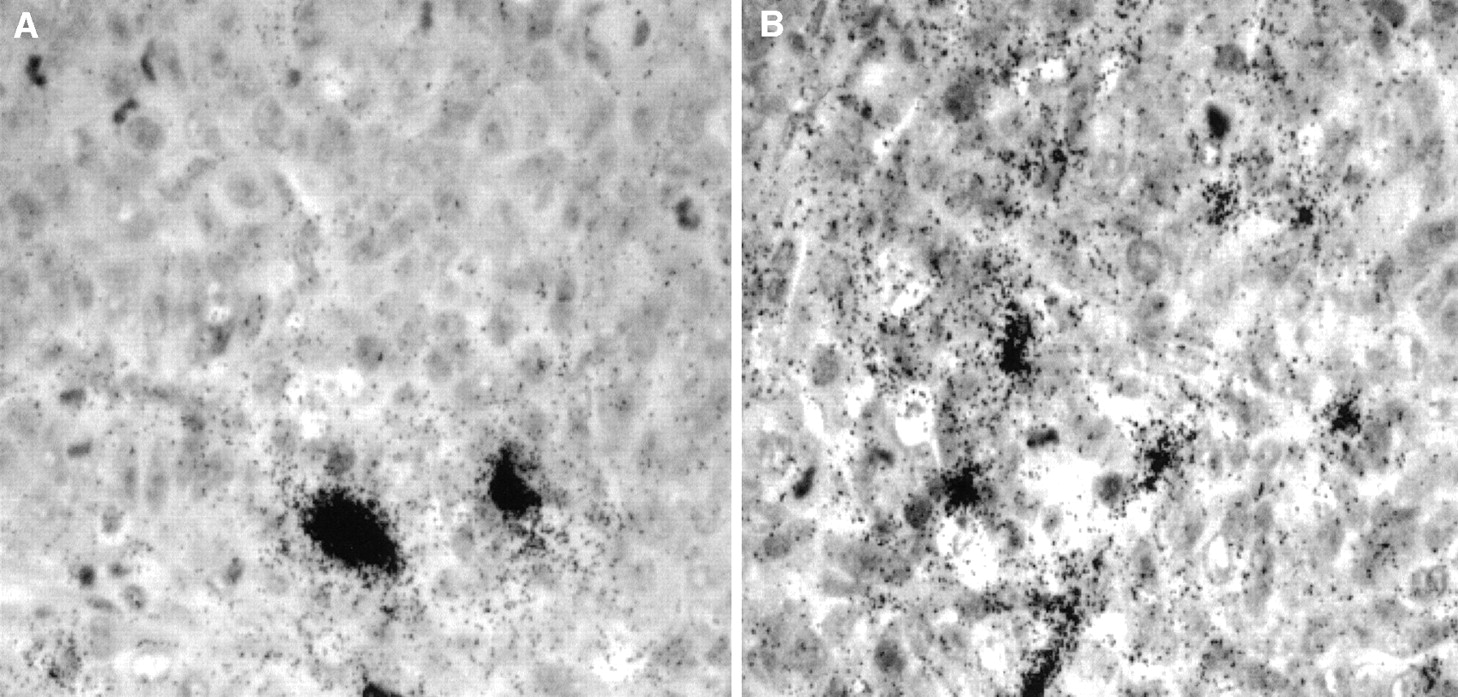
Microautoradiography of 5-μm tumor sections harvested 22 d after administration of 9.25 MBq 131I-B72.3. (A) Section of tumor from mouse treated with 131I-B72.3. (B) Section of tumor from mouse treated with 0.1 mg AP followed 3 h later by treatment with 131I-B72.3.
DISCUSSION
The growth rate of subcutaneous adenocarcinoma in animals is decreased after treatment with radiotherapeutic doses of radiolabeled antibodies. The response is usually excellent and dependent on the size of the tumor at the start of RIT and the choice of antibody. For example, a single dose of 131I-labeled CC49 produces profound tumor regression and cures when tested in subcutaneous LS174T human colorectal adenocarcinoma xenografts in athymic mice. Sixty percent of LS174T tumors treated with 18.5 MBq 131I-CC49 regress completely (23). However, in a clinical setting this outcome is never achieved. Clinical studies using B72.3 and CC49 labeled with either 131I or 90Y for the treatment of pancreatic and colorectal cancer (24–26) had only a marginal success. Although no dose-limiting, extrahematopoietic toxicity was identified, the suboptimal tumor-absorbed radiation dose estimates for 131I-CC49 suggested that further escalation of 131I-CC49 would not be useful. A similar outcome was recorded for 90Y-CC49. Even at the highest administered radioactivity, radiation doses deposited in tumor were below therapeutic threshold levels. The inevitable conclusion derived from these and other studies was unequivocal: Strategies to enhance access of radioimmunoconjugates to tumor need to be developed for effective treatment of solid tumors.
B72.3 is a prototype mAb, which recognizes a high-molecular-weight glycoprotein complex designated as TAG-72 (21) and shows significant reactivity with >85% of adenocarcinomas, including colorectal and pancreatic cancers, with only a minimal reactivity with normal tissues (27,28). Although B72.3 is excellent as a diagnostic agent, it is less effective than CC49 in animal tumor models and invariably fails in clinical RIT trials. When planning the evaluation of augmented RIT, we elected to use the less-effective, first-generation mAb B72.3 rather than CC49 to evaluate the advantages of the adjuvant treatment. These conditions, with the degree of response to RIT far less than optimal, better reflect the more difficult clinical situation encountered in RIT of adenocarcinoma.
The uneven penetration of mAb into the tumor mass, attributable in part to the impaired tumor’s VP and blood flow, results in a poor efficacy of radioimmunoconjugates in solid tumors. A discrete localization pattern of radiolabeled mAbs is evident even in small metastatic deposits. There is a good uptake of radiolabeled mAb on the edge of micrometastases, which follows the distribution of capillaries, whereas the core of the tumor remains unlabeled (29). Fractionated RIT with mAb fragments is often proposed as a way to provide a more uniform distribution of radioactivity within the tumor mass. However, a dramatic reduction of the tumor’s VP noted after just 1 administration of radioimmunoconjugates (30) suggests that repeated doses of RIT may amplify already existing problems of a nonuniform radiation dose distribution. Strategies allowing a transient increase in the VP using vasoactive agents are considered to be a feasible solution to these problems (7–14). To date, the improved therapeutic outcome resulting from modulation of the VP has not been documented. Part of the problem lies in the choice of vasoactive agents studied thus far. Cytokines, such as interleukin-2 and tumor necrosis factor-α have been shown to improve the uptake of mAbs in experimental tumor models (12,13). However, their clinical application may be difficult to realize because of toxicities observed in the course of the systemic cytokine tumor immunotherapy, such as vascular leak syndrome, hypotension, a hyperdynamic response with low systemic vascular resistance, and a host of symptoms similar to a severe infection. The mAb-cytokine constructs and fusion proteins are also tested (13,31) with the aim to target cytokines directly to the tumor site and, consequently, to minimize systemic toxicities. Increasingly, it is becoming apparent that the amount of mAbs needed to deliver effective doses of a cytokine to tumor are impractical and may be a source of additional side effects (e.g., anti-antibody response, anti-cytokine response, and so forth). To avoid shortcomings of the cytokine approach, we have examined a response-selective agonist of the human C5a anaphylatoxin as a vasoactive agent.
AP is an analog of YSFKPMPLaR, a conformationally biased, response-selective agonist of human C5a that produces marked increases in the VP in the skin of guinea pigs and hypotensive effects in rats (19,20). Structure-function studies have shown that the conformational features expressed by AP are more amenable to interacting with and activating C5a receptors (C5aR) expressed on cells responsible for these hemodynamic activities than for C5aR expressed on inflammatory PMN cells. Although used as the free peptide in this study, AP was generated by attaching the residues GCG to the N-terminal end of YSFKPMPLaR to provide a convenient way to link AP through a disulfide bridge to various carriers for the site-targeted delivery for future investigations. AP’s pharmacokinetics in mice parallel these of other agents able to interact with blood components (32). All organs except lungs clear 125I-AP with the elimination half-life of <60 min.
The effect of an enhanced VP on the tumor uptake of mAbs is known and usually manifests itself as the increase of %ID/g tumor in several tumor models (11–14,31). However, the actual augmentation of RIT in the presence of VP-modulating agents has yet to be shown. A recent report on the effect of angiotensin II-induced hypertension combined with kininase inhibition indicates that altered tumor circulation can enhance tumor targeting of radiolabeled mAb and RIT. The Tq of colorectal carcinoma xenografts increases 1.4 times when treated with RIT in combination with angiotensin II and kininase inhibitor. However, in the absence of studies with each of these agents individually, it is difficult the attribute the overall therapeutic effect to changes in the VP (11).
Our study convincingly shows an improved RIT outcome in response to the controlled changes in the tumor’s VP. A single dose of AP administered before RIT increases Tq nearly 2 times. A therapeutic protocol with 2 equal doses of 0.01 mg 24 h apart is more effective than a single dose of 0.1 mg AP. A booster dose of 0.1 mg AP results in a further delay in tumor growth, 3.5 times compared with 131I-B72.3 alone or nearly 7 times when no treatment is given. Indeed, the tumor response to a well-tolerated dose of 9.25 MBq 131I-B72.3 combined with AP produces therapeutic effects normally observed in this tumor model at the dose-limiting level of 37 MBq 131I-B72.3.
The possibility was also considered that the response of a tumor to combination radioimmunoconjugate-AP therapy may be secondary to both (a) the increased radiation dose to tumor cells caused by the enhanced VP and (b) the synergistic or additive effects of radiation and activated effector cell mechanisms. However, the absence of any tumor response in control mice treated with AP alone indicates that the immunoinflammatory effect of AP does not contribute to RIT. Additional studies are in progress to explore in greater detail the effect of AP on the tumor biology. On the basis of the currently available data, the enhanced response to RIT appears to be solely a result of the increased VP of tumor vessels.
Any therapeutic scheme involving substances that may elicit other than desired biologic responses may be difficult to translate to the clinic. The AP-augmented RIT is no exception. Of particular concern in this approach is the potential for immune responses to AP (22), especially when several administrations of the peptide are to be used. AP’s chemical structure is derived from a human C5a; therefore, if immunogenic, the responses in a rodent model would be a good indication of future problems. To date, in the studies with multiple challenges of AP administered intravenously or intradermally in normal mice, there were no detectable antibody responses. These results are encouraging and indicate that RIT with several doses of AP should not pose a problem in clinical protocols.
CONCLUSION
RIT of solid tumors can be improved significantly when judiciously selected biologic response modifiers are included in the therapy scheme. Because no blood-borne molecule can reach tumor cells without passing through the tumor’s vascular and interstitial compartments, transient increases in the VP engendered by small, nonimmunogenic peptides such as discussed in this article have a significant impact on the egress of radioimmunoconjugate into the extravascular spaces in a tumor and, consequently, improve the outcome of RIT.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (Specialized Programs of Research Excellence 1P50CA72712).
Footnotes
Received Aug. 6, 2001; revision accepted Mar. 5, 2002.
For correspondence or reprints contact: Janina Baranowska-Kortylewicz, PhD, Department of Radiation Oncology, J. Bruce Henriksen Laboratories for Cancer Research, 981050 Nebraska Medical Center, Omaha, NE 68198-1050.
E-mail: jbaranow{at}unmc.edu
REFERENCES
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Optimization of Radioimmunotherapy of Solid Tumors: Biological Impediments and Their Modulation
- Effect of Platelet-Derived Growth Factor Receptor-{beta} Inhibition with STI571 on Radioimmunotherapy
- Perspectives on Cancer Therapy with Radiolabeled Monoclonal Antibodies
- Role of polymorphonuclear leukocytes, nitric oxide synthase, and cyclooxygenase in vascular permeability changes induced by C5a agonist peptides