


Abstract
Guanylyl cyclase C (GC-C) is a transmembrane receptor expressed by human intestinal cells and primary and metastatic colorectal adenocarcinomas but not by extraintestinal tissues or tumors. The Escherichia coli heat-stable enterotoxin analog, STa (5–18), is a 14–amino acid peptide that selectively binds to the extracellular domain of GC-C with subnanomolar affinity. This study examined the utility of a radiolabeled conjugate of STa (5–18) to selectively target and image extraintestinal human colon cancer xenografts in vivo in nude mice. Methods: The STa conjugate, ethoxyethyl-mercaptoacetamidoadipoylglycylglycine-STa (5–18) (NC100586), was synthesized and labeled with 99mTc to produce 99mTc-NC100586. This compound was intravenously administered to nude mice bearing human colon cancer xenografts, and specific targeting was evaluated by biodistribution and gamma camera imaging. Results: In CD-1 nude mice, biodistribution and scintigraphic imaging analyses showed selective uptake of 99mTc-NC100586 into human colon cancer xenografts that express GC-C but not into normal tissues that do not express GC-C. Similarly, 99mTc-NC100586 injected intravenously into CD-1 nude mice with human colon cancer hepatic metastases selectively accumulated in those metastases, and ∼5-mm foci of tumor cells were visualized after ex vivo imaging of excised livers. Accumulation of 99mTc-NC100586 in human colon cancer xenografts reflected binding to GC-C because 99mTc-NC100588, an inactive analog that does not bind to GC-C, did not selectively accumulate in cancer xenografts compared with normal tissues. Also, coadministration of excess unlabeled STa (5–18) prevented accumulation of 99mTc-NC100586 in human colon cancer xenografts. Furthermore, 99mTc-NC100586 did not selectively accumulate in Lewis lung tumor xenografts, which do not express GC-C. Conclusion: This study showed that intravenously administered STa (5–18) selectively recognizes and binds to GC-C expressed by human colon cancer cells in vivo. Also shown was the ability to exploit this selective interaction to target imaging agents to extraintestinal human colon tumors in nude mice. These results suggest the utility of STa and GC-C for the development of novel targeted imaging and therapeutic agents with high specificity for metastatic colorectal tumors in humans.
Colorectal cancer is the third most common neoplasm in the United States and the third leading cause of cancer-related mortality, responsible for ∼10% of cancer-related deaths in the United States (1–5). Whereas ∼20% of patients have unresectable cancer at presentation, metastases will develop in >30% within 3 y of presumptively curative surgery, likely reflecting occult metastases undetected at staging (1–7). Staging to define the extent of extraintestinal disease determines not only prognosis but also selection of patients to receive adjuvant chemotherapy and the operative management of patients with primary and recurrent disease (5–7).
A sensitive and specific method for the accurate detection of extraintestinal disease could have a significant impact on the management of patients with colorectal cancer. Standard CT, the noninvasive imaging modality of choice for this disease, detects only metastases ≥ 5 mm (8). This sensitivity and specificity is insufficient for early detection of metastases during staging, monitoring of established disease progression, or postoperative surveillance for disease recurrence. In contrast, metabolic imaging by PET using 18F-FDG improves the detection of metastases and staging accuracy (9). Similarly, receptor-targeted nuclear imaging, which couples the superior sensitivity of radionuclide detection and the specificity of ligand–receptor interaction, has been used to detect occult micrometastases using both radiolabeled monoclonal antibodies and small peptides. The rapid clearance of small peptides relative to the extended clearance times observed for antibodies significantly reduces background radioactivity and sustains receptor targeting over intervals (1–3 h after injection) most suitable for the use of 99mTc in clinical imaging (10–15).
Guanylyl cyclase C (GC-C) is the receptor for Escherichia coli heat-stable enterotoxin (STa), an 18–amino acid peptide that is a major cause of secretory diarrhea in animals and humans (16–19). GC-C is expressed in adult humans only by intestinal epithelial cells, not by extraintestinal tissues (20–26). Of significance, expression of GC-C has been detected in all primary and metastatic colorectal tumors but not in any extraintestinal tissues or tumors examined (27–29). The apparent absolutely specific expression of GC-C outside the intestine by metastatic colorectal tumors suggests that an agent composed of STa conjugated to a radionuclide would exhibit specific uptake into those tumors with minimal uptake in normal tissues, permitting their visualization by noninvasive imaging in vivo. Such an agent could significantly affect the management of patients with colorectal cancer.
In this study, structure–function analyses of STa derivatives led to the identification of 99mTc-mercaptoacetamidoadipoylglycylglycine (N3S-adipoyl) STa (5–18) (99mTc-NC100586), which bound with high affinity to GC-C. Intravenous administration of this conjugate resulted in specific uptake into human colon cancer subcutaneous xenografts and liver metastases in nude mice, without apparent toxicity. These data suggest that STa–radionuclide conjugates selectively target GC-C on colon cancer metastases in vivo and indicate the potential utility of these conjugates as novel targeted imaging and therapeutic agents with high specificity for colorectal tumors in humans.
MATERIALS AND METHODS
Synthesis of NC100586
The requirement for correct disulfide pairing of the cysteine residues for biologically active STa (5–18) led to the development of an orthogonal strategy for selective disulfide formation. Preliminary experiments showed that the trityl, acetamidomethyl, and 4-methoxybenzyl groups sequentially formed the desired disulfide without disulfide interchange (30,31). The peptide resin, Cys(Acm)-Cys(Trt)-Glu(tBu)-Leu-Cys(Mob)-Cys(Acm)-Asn(Trt)-Pro-Ala-Cys(Trt)-Ala-Gly-Cys(Mob)-Tyr(tBu)-SASRIN, where Acm = acetamidomethyl, Trt = trityl, tBu = tert-butyl, Mob = 4-methoxybenzyl, and SASRIN = super-acid-sensitive resin, was prepared from 9-fluoroenylmethyloxycarbonyl (FMOC)-Tyr(tBu)-SASRIN resin (BACHEM Bioscience, King of Prussia, PA) using FMOC chemistry (30). All FMOC amino acids, 1,3-diisopropylcarbodiimide, N-hydroxybenzotriazole, 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate, and solvents were of reagent grade or higher. The completed peptide resin (NC100647, 10 g) was treated with trifluoroacetic acid:H2O:triisopropylsilane (97.5:1:1.5, v/v) for 30 min, followed by ether precipitation to yield 4.8 g (97% yield) of the crude linear bis-thiol (I, 1,856.7 M+Na+). The linear bis-thiol (I, 2.4 g) was air oxidized at pH 8, purified by reverse-phase high-pressure liquid chromatography (RP-HPLC), and lyophilized to yield 1.48 g (59%) of the 6–14 disulfide (II, 1,830.5 M−H−). The 6–14 disulfide (II, 1.4 g) was oxidized with iodine (5 Eq), purified by RP-HPLC, and lyophilized to yield 705 mg (55% yield) of the 6–4, 5–10 bis-disulfide (III, 1,686.9 M−H−). The bis-disulfide (III, 674 mg) was deprotected with trifluoroacetic acid:dimethyl sulfoxide:anisole (607:67:0.836, v/v) at room temperature and then concentrated on a rotary evaporator to 25% of the original volume. The residual liquid was diluted with water, purified by RP-HPLC, and lyophilized to yield 302 mg (52%) of STa (5–18). The purity, identity, and pharmacologic activity of this peptide were characterized by coinjection on isocratic RP-HPLC with authentic STa (5–18), negative electrospray mass spectroscopy (molecular weight = 1,445.0 M−H−), and GC-C binding (32), respectively. Authentic STa (5–18) was generated by digestion of STa (Sigma Chemical Co., St. Louis, MO) using porcine elastase, followed by purification by RP-HPLC.
An anhydrous N,N-dimethylformamide solution of ST (5–18) (2.9 mmol/L), ethoxyethyl (EOE)-N3S-adipoyl thiotetrafluorophenyl ester (12.4 mmol/L), and triethylamine (37 mmol/L) was stirred at room temperature for 3 h (33). The reaction was quenched by 1:3 dilution with an aqueous solution containing 0.1% trifluoroacetic acid (v/v) and 10% acetonitrile (v/v) and was immediately purified by RP-HPLC to yield 40 mg (53%) of the conjugate at 99% purity by isocratic RP-HPLC. The EOE-N3S-adipoyl STa conjugate (NC100586) was characterized by negative ion electrospray mass spectroscopy (molecular weight = 1,871.1 M−Na+) and GC-C binding analysis.
Radiolabeling of EOE-N3S-Adipoyl STa (5–18)
99mTc-NC100586 was prepared by the simultaneous deprotection and labeling of NC100586 according to the method of Kasina et al. (33). NC100586 (50 μg) was dissolved in 50 μL 80% (v/v) isopropyl alcohol in water and incubated at 75°C for 15 min with 85 μL of 0.2 mol/L HCl:glacial acetic acid (7:1; pH 1.5), 0.6 mg sodium gluconate in 150 μL water, and 150 μL fresh 99mTc-pertechnetate solution (185–259 MBq) in the presence of stannous chloride (10 μg) to yield 99mTc-NC100586. Radiochemical purity was assessed by instant thin-layer chromatography and RP-HPLC and was typically 60% before purification. The crude reaction product was purified by RP-HPLC to yield purified 99mTc-NC100586 with a typical radiochemical purity of ≥90%.
Competitive Binding Analysis
The affinity of synthetic peptides for GC-C was quantified as described previously by determining the concentration of peptide that displaced 50% of native radiolabeled STa (inhibition constant [Ki]) by competitive binding analysis (32). Briefly, increasing concentrations of synthetic ligands (0.1–500,000 nmol/L) were incubated with a fixed concentration (75 nmol/L) of 125I-STa (1–17) and 1.1 μg rat intestinal mucosal cell membrane protein in a total volume of 150 μL at 37°C for 1 h. Incubations were subjected to vacuum filtration on glass fiber filters presoaked in 0.1% polyethyleneimine to separate free from receptor-bound radioactivity. Radioactivity retained on filters was quantified to calculate free and bound radiolabeled STa. Nonspecific binding accounted for <1% of the total radioactivity bound. The concentration of unlabeled ligand that inhibited 50% of the binding by 125I-STa (1–17) (Ki) was calculated by nonlinear regression analysis using PRISM software (version 3.0; GraphPad Software, Inc., San Diego, CA).
Direct Binding Analysis
Direct binding of radiolabeled synthetic STa conjugates to GC-C was examined to determine whether chemical modification associated with radiolabeling altered the binding properties of those ligands. Briefly, RP-HPLC–purified radiolabeled conjugates (0.1–10 MBq/mL) were incubated with 6 μg rat intestinal mucosal cell membrane protein in a total volume of 150 μL at 37°C for 1 h, in the presence (nonspecific binding) and absence (total binding) of excess competing peptide (50 μmol/L STa (5–18)). Incubations were subjected to vacuum filtration on glass fiber filters presoaked in 0.1% polyethyleneimine to separate free from receptor-bound radioactivity. Radioactivity retained on filters was quantified to calculate free and bound radiolabeled STa. Specific binding was quantified as the arithmetic difference between total and nonspecifically bound radioactivity (31).
Biodistribution in CD-1 Nude Mice
99mTc-NC100586 or 99mTc-NC100588 (2 MBq, 10 ng/kg) was injected through the tail vein into female nude CD-1 mice (body weight, ∼20 g; Charles River Laboratories, Margate, UK), and at various times up to 4 h after injection the mice were euthanized and blood, liver, muscle, kidneys, lungs, and gastrointestinal tract were collected. In addition, urine and feces were collected. Tissues and carcass were weighed and counted (Wizard; Wallac, Turku, Finland). The distribution of radioactivity is expressed as percentage injected dose per tissue (%ID) or percentage injected dose per gram of tissue (%ID/g).
Biodistribution and In Vivo Imaging in Subcutaneous Tumor Model
T84 human colon carcinoma cells (0.1 mL, 1 × 108 cells per mL; American Type Culture Collection, Manassas, VA) were injected subcutaneously through a 23-gauge needle into the back of the neck of female nude CD-1 mice (body weight, ∼20 g; Charles River Laboratories). This cell line expresses GC-C at levels similar to those of human intestinal epithelial and colorectal cancer cells and is tumorigenic in nude mice (25,34–36). In all animals, 0.8- to 1.0-cm tumors (measured across 2 orthogonal axes) developed within 8–10 wk, after which the test compounds (20 MBq/mL for biodistribution studies; 200 MBq/mL for imaging studies) were injected as an intravenous bolus (0.1 mL) through the tail vein. At various times after injection, animals were euthanized and the biodistribution of test compounds were analyzed by harvesting muscle, kidneys, urine, liver, gastrointestinal tract, and tumor and obtaining a blood sample. Tissues, blood samples, and standards were weighed, and their associated radioactivity was quantified (Wizard). Each experimental point reflects data collected from ≥3 mice. The results are expressed as percentage injected dose per gram of tissue (%ID/g). 99mTc-NC100586 exhibited a short half-life in blood and rapid clearance from the body (>75% renal excretion at 60 min after injection; mean residence time, 0.82 ± 0.03 h), and consequently, tumor uptake is expressed as relative retention, calculated as the ratio of radioactivity in the tumor relative to the total radioactivity remaining in all tissues of the body at a specific time. In imaging studies, animals were euthanized at different times after injection and placed in the prone position on a gamma camera (Isocam-1; Park Medical Systems, Quebec, Canada), and whole-body planar images were acquired. Image acquisition times were typically 15–30 min or 150,000–250,000 counts (whole-body, low-energy, ultra-high-resolution collimator). Data were stored and analyzed using software from Park Medical Systems.
In Vivo Imaging of 99mTc-NC100586 in Liver Metastasis Model
Female nude CD-1 mice (body weight, ∼20 g; Charles River Laboratories) were anesthetized with halothane inhalation, a subcostal incision was made on the right lateral side, the spleen was identified, and T84 human colon carcinoma cells (1.0 mL, 2 × 106 cells per mL) were slowly injected into it. After ∼1–2 postinjection minutes, the spleen was tied off and removed and the incision was sutured. The animals recovered in a heated (37°C) chamber before being returned to their cages for 6–8 wk. Subsequently, test compounds were injected into these animals and the biodistribution was analyzed at 120 min after injection, as described above. Metastases in the excised livers were detected by histology, and their dimensions were measured across 2 orthogonal axes before the organs were placed on the gamma camera for up to 500,000 counts. Image acquisition times, data storage, and data analysis were similar to those described for the in vivo subcutaneous tumor model.
RESULTS
NC100586 Binds with High Affinity to GC-C
Previous studies showed that STa (5–18) binds to GC-C with characteristics that were nearly identical to those of native STa (37). Structure–function analyses were performed to examine the impact of various derivatization procedures, chelating groups, and metal ligands on the binding characteristics of STa (5–18). As shown previously, STa (5–18) competed with 125I-STa (1–17) for binding to GC-C in a concentration-dependent and saturable fashion, with an affinity (Ki) that was comparable with STa (1–17) (Fig. 1A; Table 1). Similarly, NC100586 also competed with 125I-STa (1–17) for binding to GC-C in a concentration-dependent and saturable fashion, with a Ki that was nearly identical to STa (1–17) (Fig. 1B; Table 1). Of significance, NC100588, an analog of NC100586 in which the 6 cysteine residues were replaced by alanines, did not compete with 125I-STa (1–17) for binding to GC-C, confirming the requirement for intrachain disulfide bridges for STa function (Fig. 1B; Table 1) (37–39). 125I-STa (1–17) and 99mTc-NC100586 both exhibited comparable specific binding to GC-C when competing with excess unlabeled STa (1–18), suggesting that radioisotopic labeling did not alter receptor binding of functional conjugates, whereas 99mTc-NC100588 exhibited no specific binding. Together, these data show that NC100586 and 99mTc-NC100586 exhibited the binding affinity and specificity required for targeting to GC-C expressed on colorectal tumors in vivo.

Competition between ST analogs and 125I-STa (1–17) for binding to GC-C. Equilibrium binding studies were conducted in presence of fixed concentrations of 125I-STa (1–17) (35–100 nmol/L) and increasing concentrations of STa (5–18) or STa (1–17) (0.1–500,000 nmol/L), and Ki values were calculated as described in Materials and Methods. (A) Competition between STa (5–18) (⧫) or STa (1–17) (▪) with 125I-STa (1–17) for binding to GC-C. (B) Competition between NC100586 (▪) or NC100588 (▴) with 125I-STa for binding to GC-C. CPM = counts per minute; M = mol/L.
Affinity of STa Analogs for GC-C
Pharmacokinetics and Biodistribution of 99mTc-NC100586
99mTc-NC100586 possesses characteristics required for specific colorectal cancer targeting in vivo, including high affinity for a receptor expressed only by metastatic colorectal cancer cells in extraintestinal sites. To examine the ability of 99mTc-NC100586 to specifically target human colorectal tumors in vivo, this conjugate or the inactive 99mTc-NC100588 was administered intravenously to non–tumor-bearing CD-1 nude mice and tissues were harvested at various times after injection for analysis of biodistribution. 99mTc-NC100586 exhibited rapid clearance from the blood, primarily by urinary excretion, with a mean residence time of 0.82 ± 0.03 h, and did not differentially accumulate in extraintestinal tissues. The inactive analog 99mTc-NC100588 may have a hepatobiliary component to its excretion, as evidenced by delayed renal excretion and higher levels of radioactivity in the liver and gastrointestinal tract (Table 2).
Biodistribution of 99mTc-NC100586 and 99mTc-NC100588 in CD-1 Nude Mice
Rapid blood clearance also was observed in CD-1 mice with subcutaneous T84 cell xenografts, in which the blood concentration of 99mTc-NC100586 decreased from an initial value of 6 %ID/g to 0.06 %ID/g within 120 min of injection. At early times, the relative retention of 99mTc-NC100586 and 99mTc-NC100588 was similar in subcutaneous colon cancer xenografts (Fig. 2). However, at subsequent times, 99mTc-NC100586 selectively accumulated in, whereas 99mTc-NC100588 rapidly cleared from, those tumors. Thus, 4 h after injection, the relative retention of 99mTc-NC100586 in tumors increased 4-fold, compared with 5 min, and was 10-fold greater than that of the inactive analog 99mTc-NC100588, which decreased 5-fold, compared with 5 min (Fig. 2). The clearance of 99mTc-NC100586 observed in CD-1 mice within 2 h was similar to that observed after 24 h with HumaSPECT (Intracel Netherlands BV, Emmen, The Netherlands), an antibody-based agent approved in Europe for imaging colorectal cancer (40). 99mTc-NC100586 administered intravenously in CD-1 nude mice specifically accumulated in subcutaneous human colon cancer xenografts that could be visualized by gamma camera scintigraphy 120 min after injection (Fig. 3A; Table 3). Similarly, 99mTc-NC100586 administered intravenously in CD-1 nude mice specifically accumulated in hepatic metastases of human colon cancer xenografts that could be visualized by ex vivo gamma camera scintigraphy of isolated livers removed at 120 min after injection (Fig. 3B; Table 3). Indeed, using this technique, human colon tumor metastases of ∼5 mm could be visualized using in vivo administration of 99mTc-NC100586 (Fig. 3).

Tumor retention of 99mTc-NC100588 and 99mTc-NC100586 in T84 subcutaneous tumors in CD-1 nude mice. Test compound (20 or 200 MBq/mL) was injected as intravenous bolus (0.1 mL) through tail vein. At various times after injection, mice were euthanized and dissected, tissues were weighed, and retained radioactivity was quantified. Data presented are average of at least 6 animals per time point and at least 2 experiments. Because of rapid clearance of labeled agent, tissue uptake is expressed as relative retention, which normalizes retained radioactivity in tissue to dose remaining in body.
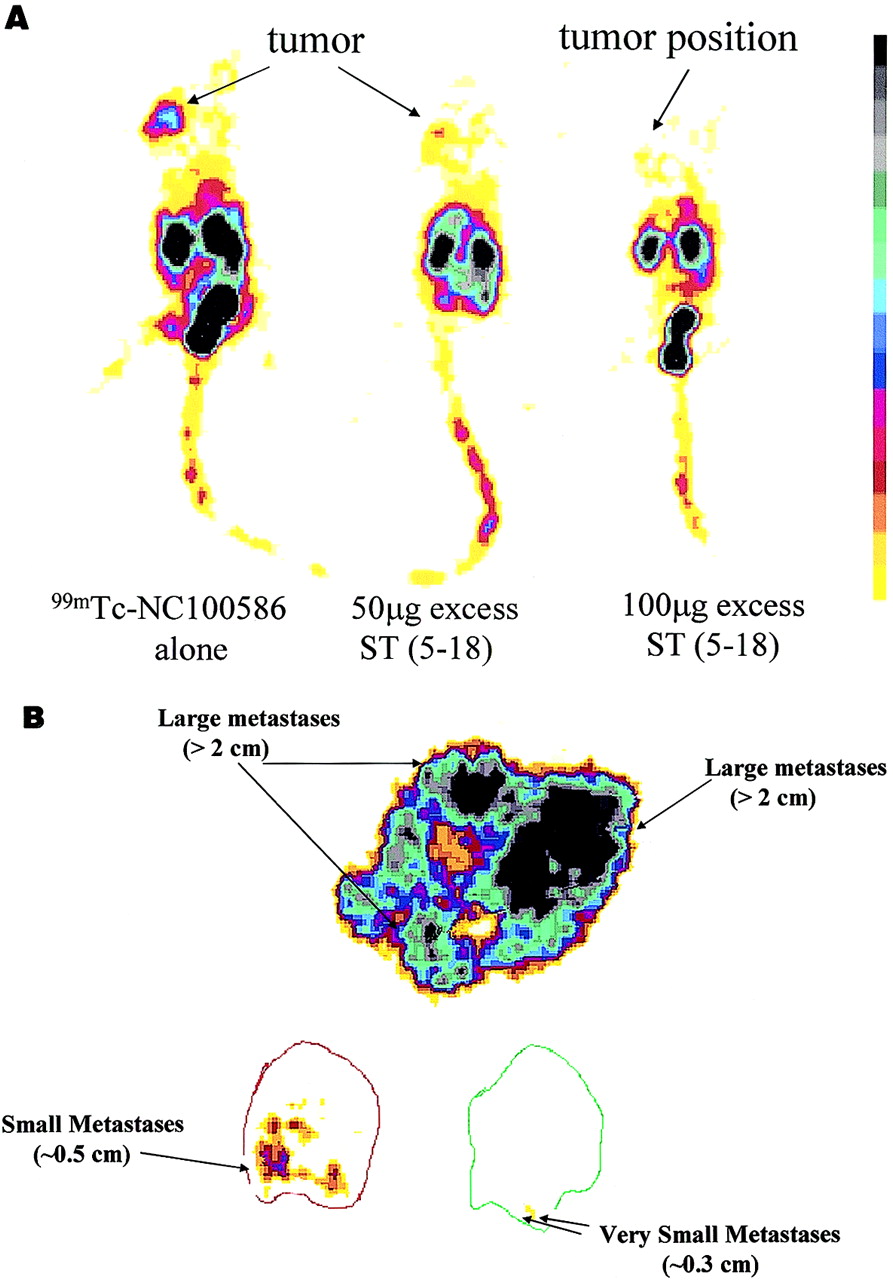
Targeting of 99mTc-NC100586 in CD-1 nude mice bearing T84 tumors quantified by gamma camera analysis. (A) Accumulation of 99mTc-NC100586 in CD-1 nude mice bearing subcutaneous T84 tumors in presence or absence of increasing doses of STa (5–18). Mice were injected with 99mTc-NC100586 (20 MBq per animal) and 50 or 100 μg STa (5–18), where indicated, as described in Materials and Methods. Planar posterior static images were collected at 120 min after injection using low-energy, ultra-high-resolution collimator. One animal from each test condition was euthanized and placed in posterior position on gamma camera (Isocam-1), and whole body planar images were acquired for 15–30 min or 150,000–250,000 counts. (B) Targeting of colon cancer metastases in liver using 99mTc-NC100586. Mice were injected with 99mTc-NC100586 (20 MBq per animal) and killed at 120 min after injection, and livers were collected as described in Materials and Methods. Planar static images were collected for up to 500,000 disintegrations using dissected tumor-bearing livers and low-energy, ultra-high-resolution collimator by gamma camera scintigraphy.
Differential Uptake of 99mTc-NC100586 into Human Colon Subcutaneous Tumors and Hepatic Metastases in CD-1 Nude Mice
Targeting of 99mTc-NC100586 to Subcutaneous Human Colon Cancer Xenografts Is Specifically Mediated by GC-C
To determine if selective tumor uptake and retention of 99mTc-NC100586 reflect an interaction with GC-C, CD-1 nude mice bearing subcutaneous human colon cancer xenografts were injected with 99mTc-NC100586 alone or with 99mTc-NC100586 and either 50 or 100 μg STa (5–18), and tumors were harvested at 120 min after injection. Injection of 100 μg STa (5–18) in a 20-g mouse represents a dose of 5 mg/kg, 500,000 times in excess of labeled peptide (10 ng/kg). Coadministration of excess STa (5–18) significantly reduced the accumulation of NC100586 in subcutaneous human colon cancer xenografts, by ∼70% (Table 4), without observable toxicity or secretory diarrhea. Similarly, coadministration of STa (5–18) with 99mTc-NC100586 reduced subcutaneous tumor uptake of the labeled conjugate in a concentration-dependent fashion, and 100 μg STa (5–18) completely eliminated differential accumulation of 99mTc-NC100586 in those tumors in vivo (Fig. 3). Furthermore, subcutaneous Lewis lung tumors, which do not express GC-C, did not selectively accumulate radioactivity on intravenous administration of 99mTc-NC100586 to host CD-1 nude mice (Table 5).
Competitive Inhibition of Accumulation of 99mTc-NC100586 in Human Colon Tumors by STa (5–18) in CD-1 Nude Mice
Accumulation of 99mTc-NC100586 in Lewis Lung Tumors and T84 Colon Tumors in CD-1 Nude Mice
DISCUSSION
This study showed that STa-based conjugates can be specifically targeted to GC-C on human colorectal tumors in vivo and used to visualize those tumors by noninvasive imaging. The GC-C–specific ligand STa (5–18) was derivatized to form NC100586 that was stably labeled with 99mTc, and this conjugate retained the ability to bind to GC-C with high affinity and specificity. Intravenously administered 99mTc-NC100586 was rapidly eliminated from the circulation, predominantly by renal excretion. After intravenous administration, 99mTc-NC100586 specifically accumulated in subcutaneous human colon cancer xenografts, but not in normal tissues, in CD-1 nude mice. In contrast, the inactive analog, 99mTc-NC100588, did not differentially accumulate in colon cancer xenografts or normal tissues in those mice. Differential accumulation of 99mTc-NC100586, compared with the inactive analog 99mTc-NC100588, in colon cancer xenografts and lack of accumulation of 99mTc-NC100586 in Lewis lung xenografts suggest that accumulation is mediated by GC-C specifically expressed by colon cancer xenografts. That coadministration of excess unlabeled STa (5–18) reduced accumulation of 99mTc-NC100586 in human colon cancer xenografts further showed that targeting of 99mTc-NC100586 to those xenografts in vivo was mediated by their differential expression of GC-C.
GC-C exhibits characteristics highly favorable for receptor-based imaging of metastatic colorectal cancer. In healthy adult humans, GC-C is expressed only by intestinal mucosal, not extraintestinal, cells (24,25,27–29). Of significance, GC-C is expressed in brush border, but not basolateral, membranes, on the luminal side of tight junctions that form the impermeable intestinal epithelial cell barrier (20–25,27–29). Thus, GC-C is normally in an anatomically privileged location, sampling the luminal environment but denied access to the vascular compartment by epithelial tight junctions. Indeed, in comparison with inactive 99mTc-NC100588, 99mTc-NC100586 did not accumulate differentially in the intestine of CD-1 nude mice in this study, supporting the suggestion that GC-C expressed by normal intestinal epithelial cells is isolated from the vascular compartment. GC-C continues to be expressed after neoplastic transformation of intestinal epithelial cells into colorectal cancer cells (24–29). To date, all human primary and metastatic colorectal tumors examined have expressed this marker, suggesting that GC-C is highly associated with the presence of colorectal cancer in extraintestinal sites (24–29). Thus, GC-C on metastatic human colorectal cancer cells appears to be a highly specific target for receptor-based imaging agents, because GC-C expressed by normal intestinal epithelial cells is inaccessible to imaging agents in the circulation. Furthermore, GC-C exhibits a high affinity for STa (∼10−10 mol/L) and a high receptor density on the surface of colorectal cancer cells (104/cell), characteristics that are particularly favorable for receptor-mediated targeting (13,17–22,27,33).
Similarly, STa (5–18) may be ideal as a targeting agent for receptor-based imaging. STa (5–18) is a 14–amino acid peptide and, consequently, exhibits favorable pharmacokinetic characteristics, including rapid clearance, compared with other receptor-based targeting agents such as monoclonal antibodies, which have clearances measured in hours and days (9–11). Also, STa (5–18) should have favorable penetration characteristics in solid tumors, reflecting its low molecular weight (<2 kDa). Furthermore, STa (5–18) has an affinity for GC-C that is 1–2 orders of magnitude higher than the affinity of all other GC-C–specific ligands previously characterized (33–35).
CT, the primary imaging modality used in colorectal cancer, detects only metastases ≥ 5 mm (8). This sensitivity and specificity are insufficient for early detection of metastases at the time of staging and for monitoring of recurrence or disease progression during postoperative surveillance. Thus, there is an unmet need for an improved method of detecting colorectal cancer metastases by noninvasive imaging with a sensitivity and specificity greater than that of CT. Intravenous administration of STa (5–18) conjugated to a radionuclide and targeted to GC-C may represent a significant improvement in noninvasive imaging of this disease. The sensitivity of gamma camera scintigraphic imaging combined with the specificity of 99mTc-NC100586 for GC-C should provide a staging method that, compared with CT, has equal or improved clinical diagnostic utility. Also, 99mTc-NC100586 is anticipated to have a greater clinical specificity than does CT, reflecting the near-absolute expression of GC-C by metastatic colorectal tumors but not by extraintestinal tissues or tumors (24–29). The increased sensitivity and specificity of STa (5–18) conjugates to detect occult disease are supported by the observation that micrometastases of ∼5 mm could be detected in livers of mice carrying hepatic human colon cancer xenografts.
CONCLUSION
The results show that STa (5–18) selectively targets and delivers an imaging agent, 99mTc-NC100586, to GC-C on the surface of metastatic colon cancer xenografts in vivo but not to the surface of Lewis lung cancer xenografts. This delivery is specifically mediated by active, but not inactive, analogs of STa (5–18) and is inhibited by excess unlabeled STa (5–18), showing that uptake of the imaging agent is dependent on differential expression of GC-C in xenografts. The rapid clearance, high binding affinity, and selective receptor targeting of NC100586 results in high sensitivity and specificity for the detection of colon cancer metastases by noninvasive imaging using this technique. Furthermore, the exquisite receptor selectivity and renal clearance observed with this peptide may make it a suitable sector for the delivery of therapeutics to colorectal cancer metastases, with significant advantages in therapeutic index.
Acknowledgments
The authors thank Edward Bacon, Christopher D.V. Black, Gemma Raymond, Tony Storey, Mark Dixon, and Harry Storflor for their insight and advice during these studies.
Footnotes
Received Jun. 15, 2001; revision accepted Dec. 3, 2001.
For correspondence or reprints contact: Henry R. Wolfe, MS, Research and Development Department, Targeted Diagnostics & Therapeutics, Inc., 1045 Andrew Dr., West Chester, PA 19380.
E-mail: wolfe{at}tdtinc.com
REFERENCES
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Preclinical Antitumor Activity and Biodistribution of a Novel Anti-GCC Antibody-Drug Conjugate in Patient-derived Xenografts
- Human GUCY2C-Targeted Chimeric Antigen Receptor (CAR)-Expressing T Cells Eliminate Colorectal Cancer Metastases
- In Vivo Imaging of Human Colorectal Cancer Using Radiolabeled Analogs of the Uroguanylin Peptide Hormone
- Receptor-mediated internalization of chelator-PNA-peptide hybridization probes for radioimaging or magnetic resonance imaging of oncogene mRNAs in tumours
- Interruption of Homologous Desensitization in Cyclic Guanosine 3',5'-Monophosphate Signaling Restores Colon Cancer Cytostasis by Bacterial Enterotoxins