


Abstract
Molecular imaging is an emerging field of study that deals with imaging of disease on a cellular or genetic level rather than on a gross level. Recent advances in this field show promise, particularly in the imaging of gene expression. This article reviews the use of nuclear medicine, magnetic resonance, and optic imaging to visualize gene expression. A review is presented of current in vitro assays for protein and gene expression and the translation of these methods into the radiologic sciences. The merging fields of molecular biology, molecular medicine, and imaging modalities may provide the means to screen active drugs in vivo, image molecular processes, and diagnose disease at a presymptomatic stage.
The ability to noninvasively monitor cell biology in vivo is the goal of molecular imaging. Tremendous advances in molecular biology have revealed the sequence, structure, and function of genes and proteins, the physicochemical properties of cellular ligands and receptors, and crucial details about the cell cycle and genetic mutations. These discoveries have led to an increased understanding of the fundamental mechanisms of disease and to the development of genetically based therapies. Research in biologic sciences combined with exploration in radiologic sciences could allow us to image the molecular basis of disease, to image responses to therapy on a molecular level, and to evaluate the effectiveness of gene transfer noninvasively.
Imaging cellular biology
Although new genes are being identified at the rate of approximately 1 per day and progress is being made in the use of new gene therapies in animal studies and clinical trials, the primary method to test for gene expression involves tissue analysis (1). Most of the current oncologic imaging technologies rely on macroscopic physical, physiologic, or metabolic changes that differentiate tumors from normal tissue rather than identifying DNA mutations or specific DNA sequences. However, as genes and their functions are further defined in vitro, progress is being made toward imaging these events (2,3). Several modalities, including SPECT, PET, MRI, and optic imaging, indicate that it is feasible to image gene expression (4–7). In recent preclinical studies, several research centers have noninvasively imaged the delivery of exogenous genes and the expression of exogenous protein. Although imaging of gene expression is currently in the realm of in vitro and preliminary animal studies, the translation to the clinical setting may be rapid because most of the marker genes that are being imaged have previously been used in gene therapy clinical trials.
The ability to image gene expression is a result of new insight into the genetic mechanisms of disease and the development of new analytic techniques to probe these genetic factors (8). These techniques include: northern blot for RNA; southern blot for DNA; western blot testing for protein expression; luminometers for luciferase marker gene detection; enzyme staining for β-galactosidase expression; fluorescent imaging of green fluorescent protein (EGFP-N1); and immunostaining for antibody or protein expression. In addition, polymerase chain reaction (PCR), which makes copies of a DNA segment, and restriction fragment length polymorphism mapping can be used for DNA fingerprinting. There also exist many peptide, antibody, and enzyme probes that confocal and fluorescent microscopic techniques exploit to image biologic processes, including mitosis, apoptosis, and necrosis.
The development of these biologic assays has had a great impact on the ability to test and understand our genetic makeup. For example, PCR can amplify the number of copies of a specific region of DNA to produce enough DNA to be adequately tested in vitro. A short chain of DNA, such as 3,000 base pairs, can be amplified about 1 million fold so that its size and nucleotide sequence can be determined. The particular stretch of DNA to be amplified, called the target sequence, is identified by a specific pair of DNA primers (oligonucleotides that are usually about 20 nucleotides in length). This technique can be used to detect, with a very high probability, disease-causing viruses or bacteria; to amplify small amounts of DNA for further analysis by DNA fingerprinting; to analyze ancient DNA from fossils; to map genomes of humans and other species; or to isolate a particular gene of interest from a tissue sample.
To qualitatively or quantitatively image gene expression noninvasively, these techniques could be adapted to in vivo systems. However, noninvasive imaging of gene expression is much more difficult than in vitro analysis, such as PCR, for 2 reasons. First, PCR thermally slices the DNA sequence, which would not be desirable in living animals. Second, molecular imaging techniques do not have PCR’s amplification capability and there are no differentiating characteristics to exploit between base pair sequences.
Gene therapy’s success depends on the accurate level, location, and duration of protein production. This must be monitored in a timely, cost effective, and minimally invasive manner. Diagnostic imaging of gene expression is essential because of the complex nature of gene transfer, including molecular biology and gene regulation, pharmacokinetics, drug delivery, and potential toxicity. Diagnostic imaging techniques could be used for gene delivery guidance, imaging vector uptake, and gene expression.
Background on Gene therapy
The goal of gene therapy is to supplement or replace the function of mutated genes with the correct genetic code. Rather than altering the disease phenotype by using agents that interact with gene products, or are themselves gene products, gene therapy can theoretically modify specific genes to correct the underlying cause of the disease. Gene therapy initially was envisioned for the treatment of inherited genetic disorders but is currently being studied in a wide range of diseases, including cancer, peripheral vascular disease, arthritis, and neurodegenerative disorders.
One of the impediments to successful gene therapy is the inefficient delivery of genes because of short in vivo half-lives, degradation by protein serum or lysosome, lack of cell-specific targeting, and low transfection efficiencies. Various gene transfer systems have been developed to penetrate the cell and deliver DNA to the nucleus where a therapeutic or marker protein can be expressed. These systems include replicant deficient viral vectors (such as adenovirus (9,10), adenoassociated virus (11), retrovirus (12,13), and herpes simplex virus (14–16)); synthetic nonviral vectors (17–23) (such as liposomes, polylysine, dendrimers, and molecular conjugates (24)); physical methods (such as the gene gun and electroporation (25)); naked DNA (26); and combinations of these various technologies (27,28).
Both ex vivo and in vivo protocols have been developed to deliver genes (29–32). For ex vivo gene therapy, the patient’s cells are extracted and the gene is inserted into the cells in vitro and then are readministered to the patient. For in vivo delivery, the gene is transferred directly to the site of interest. Typically, marker or reporter genes that do not naturally occur in the host are used to develop the vectors and to characterize their transfection efficiency or their ability to produce a foreign protein. For example, the protein produced by the β-galactosidase marker gene can be identified through enzymatic staining, the luciferase gene by a chemiluminent reaction, and EGFP-N1 by fluorometric analysis.
Adenoviral, adenoassociated, and retroviral-based systems currently account for the majority of gene therapy research because these agents are efficient carriers of genes into the cells (33). Retroviral vectors typically are replication-deficient Moloney murine leukemia viruses that are small and permit long-term expression (12,13). However, retroviral vectors replicate only in dividing cells and are expensive to manufacture because of the potential for recombination and activation. Adenoviral vectors are large (38 kilobases) and transduce nondividing cells (10,11). However, adenoviral systems are limited by a brief persistence of protein expression (usually 2 wk or less) and by the ability of the human immune system to recognize and render these viral invaders less effective upon subsequent administration (34). Furthermore, there are also limitations to the size of the gene that can be inserted into the virus and to the large-scale manufacturing of viral vectors. Adenoassociated vectors also have been developed based on a small single-stranded DNA virus and may have a lower immune response. They have the ability to transduce nondividing cells and elicit long-term expression but have a small insert size (4 kilobases) and are difficult to manufacture.
Nonviral gene delivery techniques are also being developed based on naked plasmid DNA and a variety of synthetic systems to enhance gene transfer (17–23). Although less efficient than viral vectors in terms of the amount of gene required for cell transfection, nonviral vectors are simple systems and have the advantage of a lower risk of eliciting an immune response. Naked DNA has been used in vivo and can transduce muscle tissue, but it has low transfection efficiency and is subject to hydrolysis. The nonviral gene transfection agents currently available include molecular conjugates (ligand attached to polylysine), cationic liposomes, polymers, and dendrimers. Most nonviral systems are cationic, have high amine ratios, form ionic complexes with negatively charged DNA, and can bind to the cell surface. Receptor-targeting ligands and endomolyic proteins have been added to these systems in attempts to increase transfection and specificity.
More than 3,000 patients in the United States have undergone human gene transfer in clinical trials, the majority of which have been for cancer (33). Many gene therapy protocols to date have concentrated on treatments for cancer. Though many cancers have a genetic predisposition, they all involve acquired mutations, and as they progress their cells become less differentiated and more heterogeneous with respect to the mutations they carry (35). In general, cancers have at least 1 mutation to a protooncogene (yielding an oncogene) and mutations of at least 1 to a tumor suppressor gene, allowing the cancer to proliferate. The range of different cancers encountered and the mutations they carry have led to a variety of strategies for gene therapy, namely genetic immunization, oncogene inactivation, tumor suppressor gene replacement, molecular chemotherapy, and drug resistance genes. The aim of immunopotentiation or cancer vaccines is to enhance the response of the immune system to cancers by expressing cytokines such as interleukin-2 and tumor necrosis factor-α. Oncogene inactivation may be designed to target the promoter regions of oncogenes, such as erb-2 or bcl-2, or to use antisense techniques to prevent transport and translation of the oncogenes.
Many cancers result from the abnormal function of the protein product of the p53 tumor suppressor gene (36). The p53 gene is one of the most commonly mutated genes yet identified in human cancers and its function is critical to cell cycle regulation and DNA repair. For example, mutant p53 protein may be unable to activate the transcription of molecules that control cell growth, leading to uncontrolled cell proliferation. If DNA damage has occurred, mutant p53 may not be able to arrest cell growth at the G1 checkpoint phase of the cell cycle. Augmenting the function of the p53 gene in cells that express insufficient levels of functional p53 protein could restore normal cell cycle arrest or apoptosis (programmed cell death), which may be clinically important to treating cancer.
An alternative means of killing a tumor cell is to transduce a gene coding for a toxic product, known as molecular chemotherapy or suicide gene therapy. The gene of choice is usually herpes simplex virus thymidine kinase (HSV-tk), which converts the prodrug ganciclovir (GCV) and its derivatives into toxic metabolites (15–37). Transfection of the tumor cells with the HSV-tk gene creates a biochemical difference between normal and tumor tissue that can be a target for the antiherpetic agent (38).
Nuclear Medicine Imaging of Gene Delivery and Expression
Nuclear medicine has the power to image functional and metabolic processes as well as structural morphology. Nuclear medicine has been used to characterize tumor receptor status, oncologic staging, and monitor therapeutic efficacy. For these reasons, it holds promise for imaging the emerging clinical applications in gene therapy by monitoring gene delivery and identifying protein expression. Recent studies have shown that scintigraphic imaging can offer unique information on biodistribution of the genetic vector and the extent and location of gene expression.
Radiolabeling can be used to trace the biodistribution of both viral (39) and nonviral genetic vectors (40,41) and exogenous protein expression (4–7,14,42–44). To ensure that the genetic vectors are reaching the tissue of interest, they have been labeled with radioisotopes. In Figure 1, the biodistribution of the nonviral vector, polyethyleneimine conjugated to DTPA and labeled with 111In, is shown. After tail vein injection, the polymer was rapidly cleared through the kidneys and was not specific to the subcutaneouslyimplanted tumor (rat breast 13762 NF adenocarcinoma). Radiolabeling of the vector does not show that the protein of interest is being expressed, but rather shows the location of the genetic delivery system.
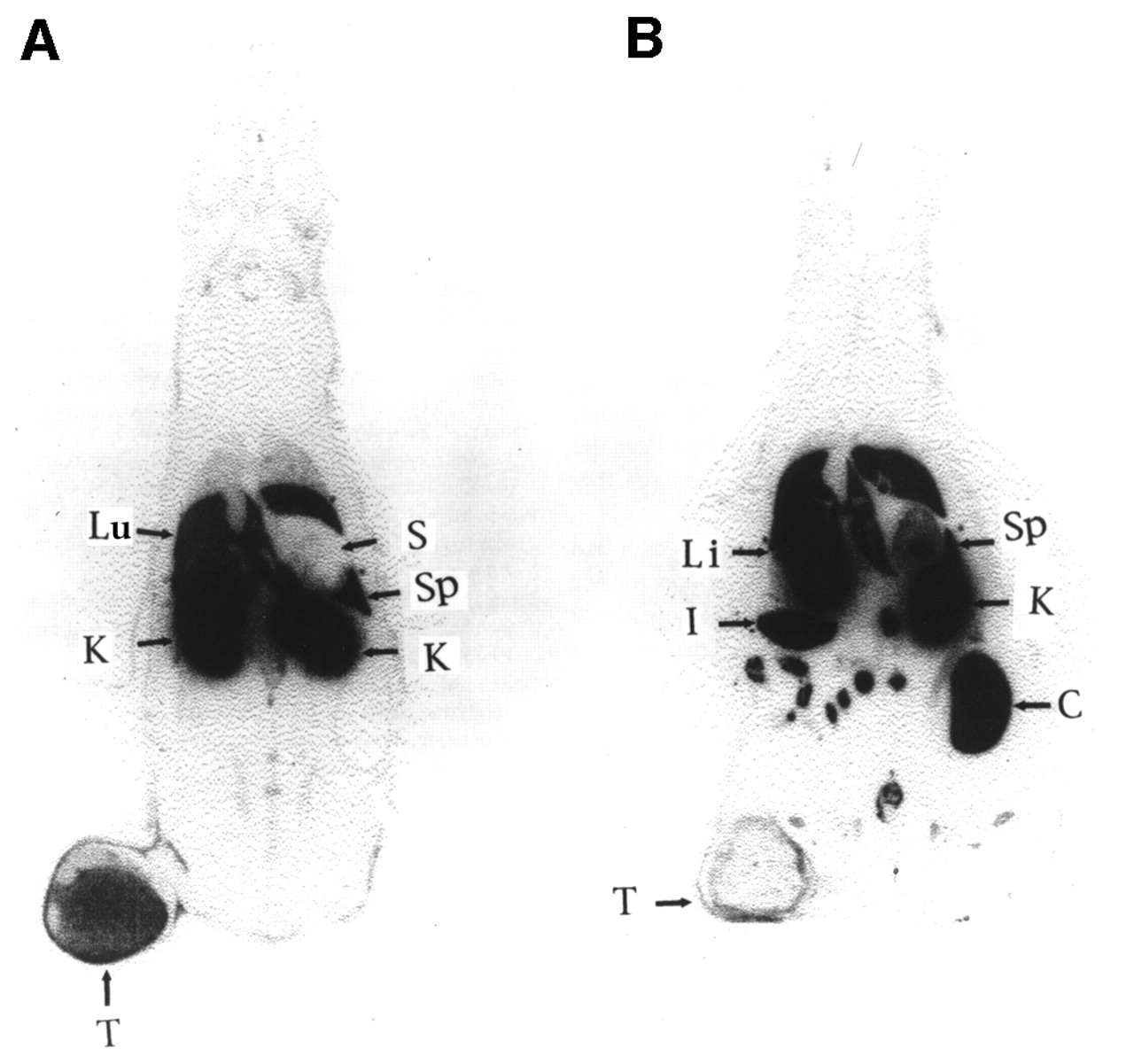
Whole-body autoradiograms (coronal section) obtained at 2 h (A) and 24 h (B) after intravenous injection of 111In-DTPA-PEI; 13762 cells (106) were implanted subcutaneously in thigh approximately 2 wk before imaging study. Lu = lung; T = tumor; K = kidney; Li = liver; I = intestine; Sp = spleen; S = stomach; C = colon.
Antibodies and ligands that target receptors that are expressed on cell surfaces can be used to understand molecular components of cells and potentially to image these components. For in vitro studies, radioimmunoassays and enzyme linked immunosorbent assay (ELISA) can be used to detect antigen-specific antibodies in a sample of body fluid or to cells in culture. For example, ELISA is used to identify individuals infected with the virus that causes AIDS (HIV) by screening for virus-specific antibodies. In vitro, the antibodies can be quite selective for the antigen. However, in vivo antibodies are impeded by biologic barriers such as substantial uptake of the antibody in the liver and poor penetration into the tumor. In fact, most radiolabeled antibodies do not yield high tumor-to-blood ratios. McKenzie and Pietersz (45) state that tumor-to-blood ratios of 2:1 and 4:1 are not uncommon in radioimmunoimaging studies.
However, antibodies, antibody fragments, and smaller ligands do play important roles in molecular imaging. For example, the epidermal growth factor receptor (EGFr) is overexpressed in a majority of cancer cells. Several studies have shown that radiolabeling the EGFr monoclonal antibody can provide images of tumors that overexpress this receptor (46,47). In addition, antibodies with improved pharmacokinetics are currently being developed by several scientists (48–50).
HSV-1-tk
One of the most exciting developments in imaging has been the use of nuclear medicine for noninvasive transgene expression imaging using herpes simplex virus type 1 thymidine kinase (HSV-1-tk) as a marker gene. The HSV-1-tk gene transfer followed by GCV treatment has been investigated as a potential gene therapy. Tjuvajev et al. (43,51,52) developed a system in which the HSV-1-tk marker gene enzyme product reacts with a radiolabeled marker substrate and converts it to a metabolite that is selectively trapped in the transduced cell. HSV-1-tk is an enzyme that is encoded in the virus. HSV-1-tk has been used as a target for nucleoside prodrug activation of treatments for herpes infection. The enzyme that is produced by the HSV-1-tk gene can phosphorylate antiherpetic agents such as GCV to its monophosphate form. Once it is phosphorylated, the substrate is unable to be transported out of the cell, so it accumulates in the transduced cell. Therefore, cells in which successful gene transfer has occurred can be distinguished from nontransfected cells.
In 1995, Tjuvajev et al. (51) showed that noninvasive imaging of HSV-1-tk gene expression was possible. They used the rat RG2 glioblastoma cell line that had been transfected in culture using the retrovirus containing the HSV-1-tk gene. Several marker substrates, including GCV, 5-iodo-2-deoxyuridine (IUDR), and 5-iodo-2-fluoro-2-deoxy-β-d-arabinofuranosyl uracil (FIAU), were evaluated as radiotracers. Both the sensitivity and selectivity of the marker substrates were tested. Sensitivity was defined by the change in substrate accumulation divided by the change in HSV-1-tk expression in transduced cells. Selectivity was defined as the sensitivity divided by the substrate accumulation caused by endogenous tk. After finding that IUDR did not exhibit adequate selectivity and that GCV had low sensitivity, the group selected FIAU for imaging HSV-1-tk positive RG2 intracerebral rat tumors. Quantitative autoradiographic images were obtained from this study. The transduced tumor is clearly identified in the left hemisphere, whereas the control RG2 tumor on the right is negative.
Several researchers (4,5,43) have since used clinical gamma cameras and SPECT to image the HSV-1-tk expression. Gambhir et al. (4) showed that [8-14C]GCV could accumulate in C6 rat glioma cells that were transfected with the HSV-1-tk gene, using autoradiography and biodistribution analysis. Using 131I-labeled FIAU, Tjuvajev et al. (43) used rats bearing subcutaneous tumors RG2 glioma or W256 mammary carcinoma cells transfected with HSV-1-tk to image gene expression. Morin et al. (5) radioiodinated another nucleoside analog, (E)-5-(2-iodovinyl)-2′-fluoro-2′-deoxyuridine, to image HSV-1-tk expression in KBALB, KBALB-LNL, and KBALB-STK murine tumor cells implanted on BALB/c mice.
PET also has been used to image HSV-1-tk expression. Several groups have synthesized 18F-labeled nucleoside analogs including acylcovir (53) and GCV (54,55) for PET imaging of HSV-tk expression. Alauddin et al. (56–58) prepared 9-[(3-18F-fluoro-1-hydroxy-2-propoxy)methyl]guanine ([18F]FHPG) for PET imaging of gene incorporation and expression in tumors. They used human colon cancer cells, HT-29, transduced with the retroviral vector G1Tk1SvNa, which showed 4 times higher uptake of the radiolabeled substrate at 1 h and up to 15 times higher at 7 h than the control (wild type) cells. In vivo studies in tumor-bearing nude mice showed that the tumor uptake of the radiotracer is 3 and 6 fold higher at 2 and 5 h, respectively, in transduced cells compared with the control cells. These results suggest that [18F]FHPG is a potential in vivo PET imaging agent for monitoring gene incorporation and expression in gene therapy of cancer.
Tjuvajev et al. (7) evaluated 124I-FIAU and showed that HSV-1-tk expression can be detected using PET. In addition, a high level of correspondence was obtained between 124I-FIAU radioactivity and independently measured HSV-1-tk expression. Levels of HSV-1-tk mRNA in the cell lines correlated to their level of sensitivity to the antiviral drug, GCV.
In addition, Srinivasan et al. (53) obtained coronal microPET images of HSV-tk expression. The group used an adenoviral vector to deliver the HSV-tk gene to C6 rat glioma cells and 18F-fluoro GCV as an imaging probe. From these studies, it is clear that PET/HSV-1-tk imaging holds promise for clinically monitoring gene incorporation and expression in gene therapy for cancer.
Receptor-Mediated Gene Imaging
Receptor-mediated imaging of gene expression uses genes to encode cell surface receptors that then can be targeted with a ligand-labeled radiotracer. For example, Raben et al. (59) used adenoviral gene transduction of human glioma D54MG cells in vitro to increase the expression of human carcinoembryonic antigen (CEA). The transduced cells exhibited high binding to 125I-labeled COL-1. In addition, the efficiency of transduction of direct intratumor injection of the adenoviral CEA vector in D54MG xenografts was determined by measuring 131I-labeled COL-1 uptake through external scintigraphic imaging.
Other reporter genes, such as the gene that produces the dopamine type 2 (D2R) receptor, also have been investigated for imaging gene expression (60). There are several substrates for D2R, including 11C-raclopride, 18F-fluoroethylspiperone (FESP), and 123I-iodobenzamine. MacLaren et al. (60) used an adenoviral viral delivery system to transfect tumor cells with a D2R reporter gene. FESP was used as a probe to target the gene expression. PET was used to image gene expression in vivo.
Another novel approach to improve molecular imaging in oncology was developed by Mandell et al. (61). They transduced cancer cells with a rNIS3 gene that facilitates iodide accumulation in follicular thyroid cells to mimic iodide uptake of the thyroid. Iodide is one of the few true “magic bullets” that allows for specific imaging of the thyroid with few side effects. In this study, the rNIS3 gene was delivered through a retrovirus to A375 human melanoma cells. Transduced and nontransduced A375 cells were inoculated intradermally and grown to 10 mm in nude mice. The rNIS-transduced tumors were visualized in vivo using γ-scintigraphy. In addition, the authors investigated the use of iodide accumulation to selectively kill transfected cells with 131I. They found that mice bearing rNIS3 transfected tumors (including A375 human melanoma, BNL 0.1ME transformed mouse liver, CT26 mouse colon carcinoma, and IGROV human ovarian adenocarcinoma cells) had a highly improved survival rate over those bearing the nontransfected tumors.
MRI of Gene Delivery
MRI is a powerful technology for noninvasive imaging of disease states. Recently, MRI has been investigated as a tool to image gene delivery by conjugating paramagnetic contrast agents to the gene delivery vector. Kayyem et al. (62) conjugated human transferrin to poly-l-lysine (PL), a cationic polymer that can form ionic complexes with negatively charged DNA. Poly-d-lysine (PDL) was attached to the paramagnetic contrast agent gadolinium diethylenetriaminepentaacetic acid (Gd-DTPA). Particles were then formed by adding varying amounts of conjugated PL to plasmid DNA, followed by the addition of Gd-DTPA-PDL to neutralize the negative DNA charge. They transfected K562 leukemia cells in vitro with the polymer/DNA complexes and analyzed the MRI T1-weighted images (repetition time/echo time = 200/13) of the transfected cells. They found that the cells transfected with the Gd-containing particles showed MRI contrast enhancement and that MRI could be used to noninvasively monitor the delivery of the polymeric gene delivery system.
More recently, de Marco et al. (63) also used MR to image gene delivery with a nonviral vector. In this study, dextran-polylysine-iron oxide/DNA particles were evaluated in vitro using a 293 embryonic kidney cell line and in vivo using adult Sprague–Dawley rats. They showed that MRI can be used to image gene delivery vectors. However, these studies imaged the delivery system rather than the expressed protein.
Initial studies have been performed using MR technology to image gene expression. To assess gene expression using MR, Weissleder et al. (64) developed a pcDNA3tyr plasmid that encodes for human tyrosinase. The tyrosinase enzyme is central in the formation of melanin that can bind with paramagnetic metals. The authors investigated whether tyrosinase expression could induce melanin production, and, in turn, if this could be imaged using γ-scintigraphy and MRI technology. A mouse fibroblast L929 cell line and a human embryonic kidney 293 cell line were transduced with the tyrosinase gene using calcium phosphate transfection protocol. Both nontransfected and mock transfected cells were used as controls. A Fontana stain with silver nitrate established the melanin production capability of these transfected cells. 111In binding studies were performed to determine the metal binding capacity of the transfected cells. The transfected 929 cells were found to have a significantly higher binding affinity than either of the control cells, and the binding was dependent on the dose of DNA. MR studies were performed on 293 transfected and mock transfected cells that were incubated in media with 5 mg iron sulfate for 3 d. Using a superconducting magnet system at 1.5 T, T1-weighted images were obtained of the cells in culture. In this study, higher signal intensity corresponded to higher expression of tyrosinase, which produced the iron-binding melanin. This study showed the feasibility of using MR for gene expression imaging, but the tyrosine induction levels are low for in vivo imaging and the gene insert size is quite large, rendering adenoviral vectors unsuitable for gene delivery.
Another approach is to use gene therapy to overexpress a cell surface receptor that can then be targeted with paramagnetic contrast agents containing a ligand for the expressed receptor. Moore et al. (65) used a gene that encoded for human transferrin receptor (hTfR) to transfect rat 9L gliosarcoma cells with 3 forms of hTfR. A protected iron containing a magnetic hTfR probe was used to show that the receptor expression could be visualized using MRI.
Optic Imaging of Gene Expression
Contag et al. (66) and Beraron et al. (67) developed a system to image luciferase reporter gene expression in vivo using bioluminescent imaging. A charged coupled device (CCD) camera (Hamamatsu Co., Tokyo, Japan) was used to detect photons emitted during the chemoluminescent reaction between the expressed luciferase and an injected or topically applied luciferin substrate. They imaged rats with B16-F0 cells that were transfected with the luciferase reporter gene and implanted subcutaneously. After the injection of luciferin substrate in dimethyl sulfoxide (DMSO), the photons emitted were clearly visible through this noninvasive imaging system. The growth of the xenografted tumor could be visualized during the 2-wk study.
Contag et al. (66) used this luminescent technology to detect reporter gene activity as a means to monitor gene expression from xenograftic tumors and to study endogenous gene regulation. The luminescence could be produced when the gene transcription was activated. They used transgenic mice with a promoter of HIV-1 that was induced by DMSO. When HIV-1 transcription was activated, the emitted photons could be imaged on the CCD camera.
Kan and Liu (68) also investigated in vivo microscopy as a tool to evaluate gene expression in living animals. Using rat 13762 NF breast cells stably transfected with EGFP-N1, the cancer cells were observed directly in vivo using video microscopy. The strength of this technology includes its ability to monitor gene expression under dynamic conditions and the potential for monitoring the metastatic phenomena associated with these green fluorescently labeled cells. With EGFP-N1 as a marker for tumor cells, in vivo microcopy could be used to monitor tumor cells in circulation, intravasculation, and cell segregation from the primary tumor. In vivo microscopy can visualize tissues within a limited depth and for a limited time because it requires anesthesia and the surgical manipulation of animals.
Imaging Endogenous Gene Expression
Imaging of endogenous genes could enhance early detection of cancer and aid in treatment decisions. For example, diagnosis of multidrug resistance (MDR) development could directly affect the treatment protocols for patients with cancer. MDR is caused by several different mechanisms. The most extensively characterized mechanism is that associated with the MDR-1 gene and its protein product, P-glycoprotein (PgP), and the multidrug resistance-associated protein (MRP). PgP and MRP are members of the ATP-binding cassette transporter family. PgP is a 170-kDa membrane glycoprotein that acts as an ATP-dependent efflux pump, reducing the intracellular accumulation of anthracyclines, Vinca alkaloids, epipodophyllotoxins, actinomycin D, taxol, and other anticancer agents. The overexpression of the MDR gene (MDR-1) is responsible for many tumors being resistant or refractory to treatment after therapy.
99mTc-sestamibi was correlated to MDR gene expression by Cordobes et al. (69). In addition, Crankshaw et al. (70) found that organotechnetium complexes, which are cationic and lipophilic, could image PgP transport. Both 99mTc-sestamibi and tetrofosmin have been used as functional probes of PgP transport activity. If these probes could be amplified and modified so that they were highly specific to a single genetic protein, then imaging of gene expression could become a reality.
Future Directions
As gene therapy continues to evolve, so will the technology to image gene delivery and expression. The resolution of imaging systems and the specificity of contrast agents have improved significantly in the last decade. In addition, the development of tumor cell markers, molecular probes, and ligands for molecular biology have created specific and detailed in vitro analysis through flow cytometry, confocal, and fluorescent microscopy. Imaging systems could be developed that would allow for the translation of tissue culture analysis systems into in vivo systems. Ultimately, the further development of new genetic markers that are readily targeted with current imaging technologies may make genetic imaging a clinical reality in the future.
Footnotes
Received Dec. 12, 2000; revision accepted May 2, 2001.
For correspondence or reprints contact: E. Edmund Kim, MD, Department of Nuclear Medicine, Box 59, The University of Texas M.D. Anderson Cancer Center, 1515 Holcombe Blvd., Houston, TX 77030-4009.
*NOTE: FOR CE CREDIT, YOU CAN ACCESS THIS ACTIVITY THROUGH THE SNM WEB SITE (http://www.snm.org/education/ce_online.html) UNTIL SEPTEMBER 2002.




{kind=link}